ОКИСЛИТЕЛЬНЫЙ СТРЕСС ПРИ ВОСПАЛЕНИИ
Воспаление (лат. in-flammare — «воспламенять») как патофизиологический феномен, или синдром, было объектом изучения с древних времен, с момента зарождения медицины. Основные признаки воспаления «rubor et tumor cum calore et dolore» впервые приведены в сохранившемся трактате «О медицине» римского доктора Корнелия Цельса (Aulus Cornelius Celsus), который жил в период с ок.
25 г. до н. э. по ок. 50 г. н. э. (рис. 1). Ощущение жара — существенная характеристика воспаления. В представлении химиков средних веков жар и горение ассоциировались с гипотетической составной частью веществ — флогистоном. Поэтому воспаление объясняли аккумуляцией флогистона в сердце, что вызывало приток крови, слизи и желчи. Флогистоновая теория воспаления была постепенно вытеснена в XVIII в. сосудистой теорией, которая на первое место ставила местные циркуляторные нарушения. Начиная с работ немецкого патолога Рудольфа Вирхова (1821—1902), многие исследователи и сегодня утверждают, что весь «облик воспаления», все его особенности, вся гамма тканевых изменений определяются сосудистой реакцией, проницаемостью сосудов микроциркуляторного русла, тяжестью его повреждения. С этих позиций воспаление рассматривается как реакция живой ткани на повреждение, которая заключается в изменениях терминального сосудистого ложа, крови и соединительной ткани и направлена на уничтожение поражающего агента и восстановление поврежденной ткани. Было доказано, что покраснение при воспалении вызвано увеличением кровотока по расширенным сосудам, а опухоль связана с экссудацией плазмы и выходом из сосудов клеток крови. Сосудистые изменения вызываются повреждающим действием флогогена, а также действием нейрогенных факторов.
Рис. 1. Шутливая иллюстрация формулы Цейса, описывающей основные признаки воспаления [1547]
Открытие в XIX в.
клеточной природы строения живых организмов, а затем и фагоцитирующих клеток привело к созданию современной концепции воспаления как местной стереотипной аварийной защитно-приспособительной сосудисто-тканевой реакции живых систем на действие патогенного раздражителя, вызывающего повреждение, и направленной на восстановление гомеостаза. С патофизиологической точки зрения воспаление относится к типовым патологическим процессам, описывающим общие закономерности ряда заболеваний разной этиологии и локализации: пневмония, перитонит, плеврит, гепатит... Главными характеристиками воспаления служат альтерация, расстройство микроциркуляции с экссудацией и пролиферация. При этом эффекторами и модуляторами воспалительного процесса являются фагоцитирующие клетки: макрофаги, моноциты, полиморфноядерные лейкоциты [141]. Бурное развитие иммунологии во второй половине XX в. привело к рассмотрению воспаления как способа реализации иммунных механизмов в критической («аварийной») ситуации, однако если данной врожденной защитной реакции недостаточно, то включается иммунное воспаление.Обращение к истории вопроса показывает, что временная концепция воспаления строится на базисе, который задается уровнем развития химии, биохимии, цитоморфологии. Основные положения современной теории были сформулированы И. И. Мечниковым еще в конце XIX в. Однако последующие сто лет концепция воспаления не единожды подвергалась корректировке. Так, Ганс Селье, развивший теорию стресса, рассматривал воспаление как местное проявление адаптационного синдрома. В XX в. воспалениие стали рассматривать как составную часть общей иммунной реакции организма с локальным проявлением.
Эти примеры свидетельствуют о том, что понятие воспаления, как и всякое субъективное понятие, отражающее сложные процессы жизнедеятельности, не является раз и навсегда установленной догмой и вызывает естественное желание исследователей вложить в концепцию воспаления современное понимание биохимических процессов, протекающих в живых системах [106].
Естественно, что развитие свободнорадикальной биологии не могло пройти мимо такого явления, как воспаление, и не включить его в список свободнорадикальных патологий.В развитии острого воспаления выделяют три фазы: альтерацию, экссудацию и пролиферацию.
На стадии альтерации (от лат. alterare — «изменять») развиваются повреждение ткани, нарушение в ней питания, а также изменяются ее структура и функции.
Первичная альтерация является результатом повреждающего воздействия воспалительного агента и зависит от свойств флогогена. Можно выделить два основных процесса включения окислительных реакций с участием АКМ на стадии первичной альтерации.
В нормальных условиях функционирования во всех клетках и мембранных структурах протекают процессы ПОЛ, которые сдерживаются на низком уровне многокомпонентной системой антиоксидантов. Важную роль в ингибировании ПОЛ играет структурная организация мембран, поэтому всякого рода повреждения структуры живой системы неизбежно сопровождаются активацией ПОЛ. Таким образом, усиление ПОЛ является универсальным ответом клеток и тканей на начальной стадии воспаления. Существует мнение, что процессы ПОЛ являются обязательным компонентом [26] и первичным медиатором стресс-реакции по Селье [20]. В эволюционном плане процессы ПОЛ, по-видимому, предшествовали эйкозаноидной регуляции [147], и поэтому продукты радикального окисления липидов мембран могут индуцировать нейрогуморальные изменения на уровне целого организма.
Конечные продукты ПОЛ (2-алкенали и 4-гидроксиалкенали) обладают высокой хемотаксической активностью: так, 4-гидроксиоктеналь усиливает хемотаксис грану- лоцитов уже в концентрациях около 10-13 М (табл. 2). Как можно видеть из таблицы, хемотаксическая активность продуктов ПОЛ значительно выше, чем активность форми- лированных олигопептидов (для К-формил^-метионил^-лейцил^-фенилаланина ED50 = 10-9 M) или лейкотриенов (для лейкотриена B4 ED50 = 10-7 М) [1639]. 4-Гидрок-
Таблица 2
Хемотаксическая активность альдегидов в отношении нейтрофилов крысы
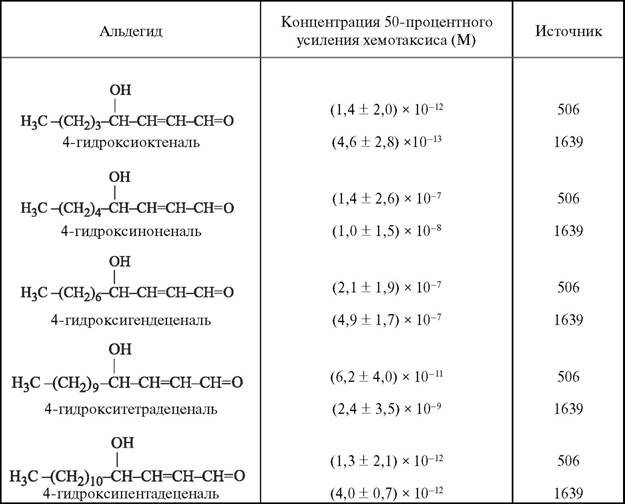
сиалкенали представляют собой биологически активные альдегиды, которые образуются при неферментативном окислении жирных кислот в клеточных мембранах или липопротеинах крови; так, при окислении арахидоновой и линолевой кислот главным продуктом является электрофильный 4-гидроксиноненаль.
В воспалительных экссудатах 4-гидроксиноненаль выявляется в микромолярных концентрациях [507], поэтому он, несомненно, участвует в хемотаксисе гранулоцитов. Продукты ПОЛ не только стимулируют направленную миграцию нейтрофилов, но могут ингибировать ее: при высоких концентрациях (10-4 М) 4-гидроксиноненаль практически полностью ингибировал направленный хемотаксис и хемокинез нейтрофилов [505, 544].Реактивные альдегиды могут взаимодействовать с SH- или NH-группами белков и тем самым изменять активность ферментов, ингибировать действие шаперонов, модифицировать структуру апобелков в липопротеинах. В результате взаимодействия 4-гид- роксиноненаля с аминокислотными остатками лизина, гистидина, цитозина образуются циклические аддукты, которые выявляются в составе белков при окислительном стрессе [128]. В концентрациях около 1 мкМ 4-гидроксиноненаль ингибировал активность тиоредоксина-1 в эндотелиоцитах посредством модификации остатка цистеина Cys73, что приводило к усилению адгезии моноцитов [666]. Ингибирование активности глицеральдегид-3-фосфатдегидрогеназы 4-гидроксиноненалем было результатом модификации остатков цистеина (Cys244, Cys281) и гистидина (His164, His327) [797]. Активация р38МАР-киназы 4-гидроксиноненалем повышала экспрессию гена и активность циклооксигеназы-2 в макрофагах и жировых клетках 3T3-L1 [1834].
В очаге воспаления продукты ПОЛ модулируют метаболическую активность фагоцитирующих клеток. Так, в концентрациях больше 1 мкМ 4-гироксиноненаль ин-
гибировал хемилюминесценцию гранулоцитов и продукцию O2 (определялась по восстановлению цитохрома с) при стимуляции клеток fMLP, ФМА и зимозаном [544, 547]. Концентрации 50-процентного ингибирования образования O2 для человеческих гранулоцитов составляли 11,6 ± 1,5 мкМ при стимуляции fMLP [544] или 27,0 мкМ при стимуляции ФМА [547]; для альвеолярных макрофагов крысы ЕД50 = 77,0 мкМ
4- гидроксиноненаля [1778]. Наибольший ингибирующий эффект достигался после 20—30 минут инкубации клеток с альдегидом, в то же время добавление 4-гидроксино- неналя непосредственно перед стимуляцией приводило к усилению метаболического «взрыва».
В низких концентрациях (от 10-11 М до 10-6 М) 4-гидроксиноненаль оказывал примирующий эффект на продукцию O2 гранулоцитами [544]. Таким образом, подобно другим липидным модуляторам, в частности простагландину Е, в низких концентрациях альдегиды оказывают провоспалительный эффект, индуцируя выход гранулоцитов в очаг воспаления и повышая их активность [144]. В то же время высокие концентрации оказывают антивоспалительное действие, снижая хемотаксис и метаболическую активность фагоцитов.Первичные продукты процессов ПОЛ также оказывают существенное влияние на активность развития воспалительной реакции. Так, гидроперекиси полиненасыщенных жирных кислот (ω-3 докозагексаеновая, ω-3 эйкозапентаеновая, арахидоновая) ингибируют адгезию моноцитов к эндотелиоцитам, стимулированным липополисахаридом, ФНО-α, ИЛ-1, ФМА; действие реализуется через ингибирование экспрессии молекул адгезии VCAM-1 и ELAM-1 [1491]. Антиатерогенный эффект богатой рыбьим жиром диеты, содержащей много полиненасыщенных жирных кислот, по-видимому, также связан с ингибированием адгезии моноцитов. Положительный эффект диеты с рыбьим жиром показан при хронических воспалительных процессах, таких как ревматоидный артрит и псориаз [336, 1491]. Интересно отметить, что действие оказывают не сами жирные кислоты, а их гидроперекиси; возможно, этим объясняется популярность у бурят омуля «с душком». Окисленно фрагментированные ω-2 ненасыщенные жирные кислоты имитируют биологические эффекты тромбоцит-активирующего фактора посредством воздействия на его рецептор [1848].
Другими источниками продукции АКМ на стадии первичной альтерации являются NO-синтаза, ксантиноксидоредуктаза, и, по-видимому, экстрацеллюлярная су- пероксиддисмутаза (Э-СОД). Образование NO-радикалов эндотелиальными клетками является важным компонентом физиологической регуляции тонуса сосудов, предупреждения тромбообразования и снижения адгезии нейтрофилов к эндотелию [147]. В эндотелиоцитах также выявляется ксантиндегидрогеназа, которая может переходить в оксидазную форму в результате окисления дисульфидных связей или ограниченного протеолиза.
Если в эндотелиоцитах в нормальных условиях на оксидазу приходится менее 1/3 суммарной активности фермента, то в очаге воспаления это значение возрастает до 2/3 и выше (величины во многом зависят от методов приготовления образцов) [1721]. Переход в оксидазную форму сопровождается резким усилением продукции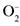 и
и 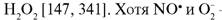 — малоактивные радикалы, однако они эффективно взаимодействуют между собой с образованием реакционного пероксинитрита:
— малоактивные радикалы, однако они эффективно взаимодействуют между собой с образованием реакционного пероксинитрита:
2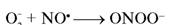
Константа скорости этой реакции определяется скоростью диффузии молекул и приближается к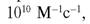 что на порядок выше константы реакции дисмутации, ката
что на порядок выше константы реакции дисмутации, ката
лизируемой СОД [147]. Ингибирование NO-радикалов в результате взаимодействия с О2 вызывает вазоконстрикцию, повышает адгезию нейтрофилов и агрегацию тромбоцитов. Кроме того, образуется пероксинитрит, обладающий высокой цитотоксичностью и индуцирующий процессы ПОЛ. Таким образом, ксантиноксидаза, находящаяся на люминальной поверхности эндотелиальных клеток капилляров, действует как молекулярный переключатель, индуцирующий синтез O2, который, в свою очередь, ингибирует NO· и способствует адгезии циркулирующих фагоцитов; в результате усилива-
ется обнаружение, поглощение и киллинг микроорганизмов ретикулоэндотелиальной системой [393]. Тонкость такой регуляции дополняется контролем со стороны Э-СОД, связанной с обращенной в просвет сосуда поверхностью эндотелиальной выстилки и лимитирующей локальную концентрацию О2. На модели развития отека лапы у крыс после введения каррагинана показано, что ингибирование конститутивной NO-синтазы снижало развитие отека в первые четыре часа, в то время как угнетение индуцибель- ной NO-синтазы снижало развитие отека на более поздней стадии (4—10 часов) [1450].
Развитие окислительного стресса при воспалении не только индуцирует поступление фагоцитов в очаг воспаления (см. табл. 2), но и модулирует их рецепторные свойства, а также вызывает так называемую активацию эндотелия — регулируемое изменение фенотипа эндотелиальных клеток, характеризующееся повышением экспрессии на их поверхности молекул адгезии и других белков, участвующих в межклеточных взаимодействиях. Так, H2O2 индуцировала экспрессию Fс-рецепторов (CD64, CD32, CD16) для иммуноглобулинов на поверхности нейтрофилов, при этом содержание рецепторов на азурофильных гранулах и секреторных везикулах снижалось, также снижалось содержание С-рецепторов (CD35,CD11b/CD18) для белков системы комплемента на цитоплазматической мембране; в отличие от перекиси водорода O2 индуцировал экспрессию С-рецепторов на поверхности клеток [1522]. Во многих работах показано, что характерное для воспалительной реакции усиление «липкости» эндотелия опосредуется активными формами кислорода, в том числе супероксид-анионом, генерируемым в результате активации NAD(P)H-оксидаз медиаторами воспаления: под их действием на эндотелиальных клетках возрастает экспрессия адгезивных молекул ICAM-1, VCAM-1, селектинов [1385].
Если резюмировать основные события, происходящие на начальной стадии воспаления, то можно сказать, что развитие окислительного стресса вызывает вазоконстрик- цию, усиливает агрегацию тромбоцитов и адгезию нейтрофилов к эндотелию; продукты ПОЛ стимулируют миграцию в ткани гранулоцитов и моноцитов, а также вызывают нейроэндокринные изменения на уровне целого организма.
При развитии воспалительной реакции выделяется фаза вторичной альтерации, которая является следствием воздействия на соединительную ткань и микрососуды высвобождающихся из клеток лизосомальных ферментов и АКМ. Вторичная альтерация определяется преимущественно состоянием общего и местного иммунитета, и прежде всего — активностью фагоцитирующих клеток. В экспериментальных исследованиях показано значительное снижение выраженности альтерации у животных с предварительно вызванной лейкопенией. Синтез АКМ и усиление свободнорадикальных окислительных процессов при альтерации вызывает повреждение клеток и тканей, повышается проницаемость мембран. Наработка органических перекисей вызывает закисление среды в очаге воспаления.
Если на стадии первичной альтерации наблюдается ингибирование NO-радикалов, что приводит к вазоконстрикции, то на более поздней стадии под действием цитокинов и бактериальных липополисахаридов в клетках активируется индуцибельная NO-синтаза. Активность ее в 100—1000 раз выше активности конститутивной NO-синтазы и не зависит от ионов Са2+. Максимальная скорость синтеза NO-радикалов макрофагами грызунов — 100 нмоль/час на мг клеточного белка [471], в пересчете на 1 клетку это составляет около 10 миллионов молекул NO· в секунду. Провоспалительные цитокины повышают продукцию NO· не только посредством усиления синтеза индуцибельной NO-синтазы, но также и посредством повышения экспрессии диметиларгинин диме- тиламиногидролазы, отвечающей за метаболизм эндогенного ингибитора NO-синтаз — асимметричного диметиларгинина [48, 1663].
Впервые появление оксидов азота (продуктов окисления NO·) как медиаторов воспалительного процесса было показано Tannenbaum et al. [1603] в 1978 г. Изучая содержание неорганических нитритов и нитратов в выделениях человека, исследователи показали, что из организма человека выделяется значительно больше нитрата, чем поступает с пищей, при этом экскреция значительно возрастала при инфекционных и других воспалительных процессах. В 1985 г. макрофаги были идентифицированы как эффективный источник эндогенных нитрата и нитрита, предшественником которых выступал L-аргинин. Дальнейшие исследования показали, что NO· образуется в ферментативной реакции с NO-синтазой, экспрессия которой не ограничена только иммунокомпетентными клетками — макрофагами и гранулоцитами. В эндотелиоцитах при действии липополисахарида или цитокинов, таких как фактор некроза опухоли-α, помимо конститутивной формы фермента возможна экспрессия индуцибельной NO- синтазы [887]. Кроме того, в эндотелиоцитах комбинация интерферона-γ с фактором некроза опухоли-α или интерлейкином-ф значительно усиливает активность конститутивной NO-синтазы посредством увеличения внутриклеточного содержания тетра- гидробиоптерина, низкая концентрация которого в нормальных условиях ограничивает наработку NO· [1430].
In vivo инъекция Corynebacterium parvum вызывает воспаление в печени и приводит к усилению активности NO-синтазы в гепатоцитах в первые 3—7 дней после введения, максимум активности наблюдается на 4 день [333]; концентрация NO3 в сыворотке и моче возрастает в первые 3 дня, затем снижается [648]. Индукция NO-синтазы происходит в ответ не только на бактериальные стимулы или цитокины, но также и на вирусы, в частности вирус гепатита [1039]. Высокий уровень генерации NO-радикалов (локальная концентрация NO· может возрастать более чем в 100 раз, концентрация NO3 в крови повышается в 15 раз [1040]) в результате активации индуцибельной NO-синтазы приводит к неконтролируемой вазодилатации в очаге воспаления, а следовательно — к усилению кровоснабжения, необходимого для удаления токсических продуктов, и в дальнейшем — для поступления нужных для репарации компонентов.
Помимо вазодилатации, высокие концентрации NO-радикалов в очаге воспаления оказывают микробицидное, туморицидное, антипролиферативное действие и модулируют развитие воспалительной реакции [144, 341]. NO· влияет на метаболическую и секреторную активность макрофагов [242], в частности, ингибирует 5-липоксигеназу и NADPH-оксидазу [147] и тем самым снижает синтез медиаторов воспаления — лей- котриенов и О2, а также стимулирует активность циклооксигеназы и усиливает синтез простагландина E2 [1161]. Вместе с тем обнаружено, что клетки Купфера синтезируют значительно больше простагландина E2 и интерлейкина-6 в ответ на интерферон-γ и липополисахарид, если NO-синтаза заингибирована [1550]. Показано супрессивное действие оксида азота на активацию нейтрофилов (реорганизацию актина и хемотаксис) [1720] и их цитотоксичность: так, эндотелиоциты микрососудов легких, синтезирующие больше NO· в ответ на комбинацию «интерферон-γ + липополисахарид», чем дермальные, менее подвержены повреждающему действию со стороны ФМА-стимули- рованных гранулоцитов [1183]. NO-радикалы могут также влиять на жизнеспособность макрофагов [243, 1458] и гранулоцитов [1720] путем индукции их апоптоза. Спектр биологического действия NO-радикалов очень широк: они снижают адгезию и хемотаксис гранулоцитов, агрегацию тромбоцитов, ингибируют пролиферацию лимфоцитов и гладкомышечных клеток, модулируют синтез инсулина и тиреоидных гормонов и т. д. [147, 858].
Как правило, воспалительный процесс носит локальный характер. Вместе с тем возможны системные воспалительные реакции организма, которые, в частности, наблюдаются после внутривенного введения экспериментальным животным бактериальных липополисахаридов или цитокинов, таких как интерлейкин-1 или фактор некроза опухолей, эти ситуации моделируют клинические состояния сепсиса. При сепсисе активация NO-синтазы и избыточная продукция NO· приводит к снижению тонуса сосудов и падению системного давления, что является причиной коллапса сосудов и шока [1230]. Через 3—6 часов после введения липополисахарида или фактора некроза опухоли у животных повышается экспрессия мРНК индуцибельной NO-синтазы, максимум индукции наступает через 6 часов. Синтез NO· наблюдается в макрофагах, гепатоцитах, кардиомиоцитах и других клетках. Максимальные концентрации NO· и NO- в крови достигаются через 12 часов [1223]. В клинических условиях индукция NO-синтазы и развитие гипотензивного состояния возникает также при проведении иммунотерапии с использованием цитокинов, в частности интерлейкина-2 [754]. Ингибиторы синтеза NO· препятствуют развитию гипотензивных состояний у экспериментальных животных после введения липополисахарида или фактора некроза опухоли [1223]. Положительный эффект применения ингибиторов NO· с целью повышения артериального давления и поддержания тонуса сосудов получен также у больных с сепсисом [1316]. Вместе с тем экспериментальные исследования и клинические наблюдения показывают, что применение ингибиторов NO-синтазы не всегда оправдано, так как эти препараты усиливают свободнорадикальное повреждение миокарда, печени и тонкого кишечника [1223, 1316].
Дегрануляция и высвобождение МПО при стимуляции фагоцитов, прежде всего нейтрофилов, приводит к усилению процессов ПОЛ и индукции образования эйкоза- ноидов. Этот процесс, по-видимому, не связан прямо с синтезом HOCl или других ги- погалогенитов, а опосредуется через окисление оксидов азота [1836]. Перитонит, индуцированный введением зимозана, у нокаутированных по МПО мышей сопровождался значительно менее выраженным накоплением F2-изопростанов и продуктов окисления линолевой кислоты. Так как активация гранулоцитов достаточно быстрый процесс, то это позволяет рассматривать МПО как один из главных ферментов, ответственных за усиление процессов ПОЛ и синтез провоспалительных эйкозаноидов на начальной стадии развития воспалительного процесса.
Экссудация (от лат. exsudo — «потеть»). Изменение микроциркуляции и нарушение проницаемости мембран эндотелия в результате их повреждения приводит к поступлению плазмы в мышечный слой. При малых повреждениях наблюдается преимущественная экссудация жидкости и низкомолекулярных соединений, усиление деструкции приводит к выходу высокомолекулярных соединений вплоть до клеток. Экссудация и развитие отека во многом определяются состоянием микроциркуляторного русла и эффективностью кровотока. Эндотелиоциты микрососудов чувствительны к повреждающему действию АКМ, особенно Н2О2, что определяется низким содержанием внутриклеточной каталазы в данных клетках [1509]. Ингибиторы NO-синтазы вызывают вазоконстрикцию и ингибируют развитие экссудативных процессов при воспалении [1340], аналогичное действие оказывают глюкокортикоиды, которые, как известно, снижают синтез NO· [1724]. Вместе с тем ингибирование образования NO-радикалов усиливает адгезию нейтрофилов к эндотелию и их повреждающее действие; NO· также защищает эндотелиоциты от токсического действия окисленных липопротеинов и снижает их апоптоз.
В развитии отека при воспалении существенную роль могут играть тучные клетки. Их активация приводит к высвобождению гистамина и тромбоцит-активирующего фактора, в результате чего усиливаются роллинг и адгезия лейкоцитов. С помощью флуоресцентной микроскопии небольших участков венул было показано, что экзогенные NO-радикалы слабо влияют на гистамин-индуцированные роллинг и адгезию, вызванные тромбоцит-активирующим фактором, однако NO· ингибировал данные процессы, если они вызывались активацией тучных клеток [632].
На экспериментальной модели артрита, индуцированного у мышей перхроматом калия (K3QO8), было показано, что ингибиторы NADPH-оксидазы (дифенилиодоний и стауроспорин) в фагоцитирующих клетках снижали развитие воспалительного отека [1144]. Аналогичный противовоспалительный эффект наблюдается при действии антибиотиков, способных ингибировать NADPH-оксидазу [1670]. Поэтому экссудация во многом зависит от интенсивности и соотношения образования разных форм АКМ, индуцирующих повреждение эндотелиоцитов, а также состояния антиоксидантной защиты эндотелия.
Эффективными модуляторами синтеза АКМ в очаге воспаления являются цитокины, среди которых обладающие провоспалительным действием (Г-КСФ, ГМ-КСФ,
ФНО-α, интерферон-γ, ИЛ-1, ИЛ-2, ИЛ-6, ИЛ-8 и фактор активации тромбоцитов) усиливают продукцию радикалов O2- и NO·, в то время как антивоспалительные цитокины (росттрансформирующие факторы, ИЛ-4, ИЛ-10, ИЛ-13) снижают (табл. 3). Как правило, наиболее полный метаболический ответ фагоцитирующих клеток наблюдается при воздействии комбинации двух стимулов, например, интерферона-γ и бактериально-
Таблица 3
Действие цитокинов на продукцию АКМ фагоцитирующими клетками
| Фактор | Эффект | Источник |
| Г-КСФ | Примирует гранулоциты человека | 293, 867 |
| При адгезии усиливает синтез O2 | 1199 | |
| М-КСФ | Примирует макрофаги, | 1768 |
| гранулоциты | 866 | |
| и моноциты человека | 1831 | |
| ГМ-КСФ | Примирует гранулоциты | 293 |
| и моноциты человека | 900,1831 | |
| При адгезии усиливает синтез O2 | 1199 | |
| Инсулиноподобный фактор роста-1 | Примирует нейтрофилы человека | 339 |
| Росттрансформирующие факторы β1, β2, β3 | Ингибируют индукцию NO-синтазы | 548 |
| ФНО-α | Индуцирует NO-синтазу | 357, 554 |
| Примирует нейтрофилы | 291, 1673 | |
| и моноциты человека | 900 | |
| Интерферон-γ | Индуцирует NO-синтазу | 554, 887 |
| Примирует нейтрофилы | 1611 | |
| и макрофаги | 1762 | |
| ИЛ-1 | Индуцирует NO-синтазу | 357,1027 |
| Примирует гранулоциты, моноциты | 88 | |
| ИЛ-2 | Примирует нейтрофилы человека | 601 |
| Индуцирует синтез NO· в организме | 754 | |
| человека | ||
| ИЛ-3 | Примирует эозинофилы | 1677 |
| и моноциты человека | 1831 | |
| ИЛ-4 | Ингибирует NO-синтазу, | 249, 420 |
| Снижает синтез O2 моноцитами | 227 | |
| ИЛ-5 | Примирует эозинофилы человека | 1677 |
| ИЛ-6 | Примирует нейтрофилы | 356 |
| и моноциты человека | 885 | |
| ИЛ-8 | Примирует нейтрофилы человека | 948,1782 |
| ИЛ-10 | Ингибирует индукцию NO-синтазы, | 420, 503 |
| но усиливает образование NO· | 492 | |
| ИЛ-12 | Индуцирует и потенцирует синтез NO· | 1757 |
| ИЛ-13 | Ингибирует индукцию NO-синтазы | 1467 |
| и образование NO· | 347 | |
| Фактор активации | Примирует гранулоциты | 1597 |
| тромбоцитов | и эозинофилы человека | 344 |
| Примирует макрофаги печени | 310 | |
| При адгезии усиливает синтез O2, | 1520 | |
| индуцирует синтез NO · | 144,1475 |
го липополисахарида или ИЛ-Ια и интерферона-γ, при этом роль первого примирую- щего клетки стимула обычно выполняют цитокины. Механизмы действия цитокинов во многом неясны. Так, показано, что эффект Г-КСФ, ГМ-КСФ и ИЛ-8 реализуется через активацию G-белков в нейтрофилах человека [293, 948], однако пути перевода G-белков в активное состояние для них различаются; ФНО-α и интерферон-γ прямо индуцируют экспрессию генов, кодирующих синтез тяжелой цепи (gp91phox) цитохрома b558 и цитозольных факторов p47phox и p67phox NADPH-оксидазной системы [697]. ИЛ-1 и ФНО-α активируют ядерный фактор транскрипции NF-κΒ, что вызывает экспрессию ряда генов, и в том числе кодирующих РНК индуцибельной NO-синтазы, при этом их эффект проявляется не только в отношении фагоцитирующих клеток: они также усиливают синтез фермента в гепатоцитах, фибробластах [251], β-клетках поджелудочной железы [490] и клетках щитовидной железы [858] и, возможно, в большинстве фенотипов соматических клеток [753]. Кроме того, в макрофагах, фибробластах и эндотелиальных клетках интер- ферон-γ и ФНО-α индуцируют синтез de novo тетрагидробиоптерина, являющегося кофактором NO-синтаз [1319].
Возможно, что регуляторное действие цитокинов при воспалении во многом реализуется через активацию наработки АКМ. Вместе с тем сами АКМ индуцируют синтез некоторых цитокинов. Так, было показано, что образование ИЛ-8 в цельной крови человека, моноцитах, фибробластах и эпителиальных клетках ингибируется антиоксидантами и усиливается активацией эндогенных механизмов синтеза АКМ или добавлением экзогенных АКМ [1400], аналогичное повышение синтеза монокинов выявлено в моноцитах [990]; индуцированный частицами кварца синтез ФНО-α, макрофагального воспалительного белка-2 и моноцитарного хемотаксического пептида-1 супрес- сировался антиоксидантами [301]. Перекись водорода в низких концентрациях (10-5 М) усиливала продукцию Т-лимфоцитами ИЛ-2 [88]. Действие АКМ реализуется через активацию ядерного фактора транскрипции NF-κΒ, который контролирует синтез ряда иммунорегуляторных цитокинов, молекул адгезии и белков острой фазы.
Состояние внутренней среды организма также оказывает существенное влияние на метаболическую активность фагоцитирующих клеток: так, высокий уровень глюкозы в крови при диабете [758] или урата при подагре [1621] усиливают продукцию АКМ полиморфноядерными лейкоцитами и, соответственно, провоцируют развитие окислительного стресса в очаге воспаления.
Еще по теме ОКИСЛИТЕЛЬНЫЙ СТРЕСС ПРИ ВОСПАЛЕНИИ:
- Механизмы развития окислительного стресса при диабете типа II
- Окислительный стресси дисфункция β-клеток при сахарном диабете типа II
- Окислительный стресс и инсулинрезистентность
- Окислительный стресс и диабетические ангиопатии
- Психология травматического стресса. Методологические различия при определении стресса и травматического стресса
- Меньщикова Е. Б.. Окислительный стресс: Патологические состояния и заболевания / Е. Б. Меньщикова, Н. К. Зенков, В. З. Ланкин, И. А. Бондарь, В. А. Труфакин.— Новосибирск,2008. - 284 с., 2008
- Воспаления зрительного нерва
- Воспаление слезного мешка
- Окислительная модификация ЛНП и атеросклероз
- Клиника Воспаления слезных органов и глазницы
- Экспериментальные модели окислительного поражения легких у животных