Окислительный стресси дисфункция β-клеток при сахарном диабете типа II
Ожирение — основной фактор риска для манифестации сахарного диабета типа II [693]. Развитие ожирения также связывают с инсулинрезистентностью [1057]. Однако у большинства людей с ожирением диабет не развивается благодаря компенсаторной гипер- инсулинемии, позволяющей преодолевать барьер инсулинрезистентности: так, у больных с ожирением, не страдающих сахарным диабетом, увеличивается масса β-клеток [910, 1417].
В то же время у больных сахарным диабетом типа II по данным аутопсий отмечается снижение массы β-клеток на 40—60 % [473, 910]. Показано, что наибольшая масса инсулярного аппарата отмечается у лиц без сахарного диабета с ожирением, тогда как минимальная масса β-клеток отмечена у пациентов с сахарным диабетом типа II без ожирения. По данным Kloppel et al. [910], у больных диабетом типа II масса β-клеток снижена на 50 % в сравнении с массой инсулярного аппарата у лиц без диабета, страдающих ожирением, и только слегка снижена по сравнению с худыми, не страдающими диабетом субъектами. Эти данные свидетельствуют о том, что для людей с сахарным диабетом II типа характерно снижение резерва β-клеток для адекватного обеспечения потребности в инсулине.Масса β-клеток регулируется балансом между клеточной репликацией и апоптозом и пополняется за счет неогенеза островков из эпителия экзокринных протоков поджелудочной железы. Не существует единого мнения, почему уменьшается масса β-клеток, приводя, в конечном итоге, к снижению секреции инсулина при сахарном диабете типа II [910, 1444], однако недавно были опубликованы убедительные данные, свидетельствующие, что именно нарастание апоптоза, а не снижение онтогенеза или репликации, приводит к снижению массы β-клеток при данном заболевании. Авторы сравнивали аутопсийный материал у тучных и худых индивидуумов с наличием и отсутствием диабета или нарушенной толерантности к углеводам.
Было показано, что у тучных субъектов без диабета масса β-клеток в 1,5 раза выше, чем у худых лиц, не страдающих диабетом. Для тучных пациентов с нарушением толерантности к глюкозе и сахарным диабетом типа II было характерно снижение массы β-клеток соответственно на 40 % и 63 % по сравнению субъектами, страдающими ожирением, но не диабетом, а для худых пациентов с диабетом — на 41 % по сравнению с худыми лицами без диабета. При этом было показано, что неогенез инсулярного аппарата, возрастающий по мере увеличения веса, сравним во всех группах, а частота апоптоза β-клеток в 10 раз больше у худых и в 3 раза — у тучных больных диабетом, чем у здоровых лиц соответствующих групп сравнения [399]. Инкубация островков Лангерганса, выделенных у больных диабетом типа II, с метформином приводила не только к стимуляции работы инсулярного аппарата и нормализации показателей редокс-баланса, но и к угнетению апоптоза и ингибированию каспазы-3 и каспазы-8 [1096].Исследования, показавшие, что длительная экспозиция высоких концентраций глюкозы приводит к усилению апоптоза β-клеток и снижению секреторных возможностей инсулярного аппарата, позволили ввести новое понятие — «глюкотоксичность» («глюкозотоксичность») [1335]. Длительная, повторяющаяся гипергликемия обуславливает обратимое снижение чувствительности β-клеток к стимуляции глюкозой, которая восстанавливается после нормализации уровня глюкозы в крови. Однако хроническая гипергликемия в конечном итоге приводит к истощению β-клеток за счет снижения в них запасов инсулина, вследствие чего становится невозможной адекватная секреция последнего, даже если и происходит восстановление чувствительности β-клеток к уровню глюкозы. Важное различие между истощением β-клеток и глюкотоксичностью заключается в том, что в первом случае не отмечено дефекта в синтезе инсулина и клеточная функция полностью восстанавливается после отдыха β-клеток. Глюкотоксичность, напротив, приводит к необратимым повреждениям в клеточных компонентах, участвующих в процессе синтеза, накопления и секреции инсулина.
Концепция глюкотоксичности в гораздо большей степени применима для диабета типа II, нежели для диабета типа I, поскольку, как отмечалось выше, у пациентов с диабетом типа II β-клетки остаются функциональными в течение многих лет после возникновения заболевания. При этом медикаментозное воздействие поддерживает оптимальный уровень глюкозы натощак, в то время как ее постпрандиальное содержание, как правило, повышено, и такие пациенты подвергаются двойному риску, поскольку функция их β-клеток снижается не только этиологически, в результате причин, вызывающих заболевание, но и патогенетически, под действием обусловленных этим заболеванием аномальных концентраций глюкозы [1417]. Было показано, что глюкотоксичность влияет на все важнейшие этапы секреции инсулина, начиная от экспрессии гена синтеза инсулина и заканчивая поступлением инсулина в кровь, в том числе угнетение процессов трансляции в синтезе инсулина, подавление экспрессии гена глюкокиназы, снижение митохондриальной функции, нарушение механизмов экзоцитоза и, наконец, ускорение апоптоза [399, 1074].
Одним из механизмов, опосредующих глюкозо-индуцированную дисфункцию β- клеток, может служить активация белка-разобщителя UCP2 [374, 433]. Гипергликемия, увеличивая продукцию доноров электронов NADH и FADH2 в цикле Кребса, тем самым способствует повышению митохондриального трансмембранного потенциала (см. рис. 15; рис. 17), что, в свою очередь, приводит к угнетению транспорта электронов на комплексе III, возрастанию времени полужизни радикальных интермедиатов коэнзима Q и гиперпродукции О-. Показано [933], что индуцированный гипергликемией рост генерации супероксид-аниона в митохондриях сопровождается активацией опосредованного разобщителем UCP2 поступления протонов, снижением трансмембранного потенциала и уровня ATP, что служит причиной нарушения ATP-зависимого процесса глюкозо-стимулированной секреции инсулина.
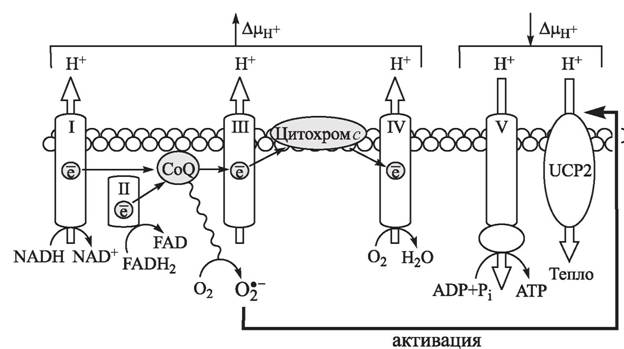
Рис.
17. Влияние гипергликемии на функционирование электронтранспортной цепи митохондрий β-клеток поджелудочной железы [374]Federici et al. [595] продемонстрировали, что в культуре человеческих панкреатических островков, инкубируемых с высокими концентрациями глюкозы, отмечается гиперэкспрессия проапоптотических белков Bad, Bid и Bik, тогда как экспрессия анти- апоптотического Bcl-2 не изменяется, что свидетельствует о нарушении баланса в сторону апоптоза и гибели β-клеток. β-Клетки продуцируют ИЛ-ф в ответ на хроническую гипергликемию, что, в свою очередь, приводит к активации апоптозного пути (активация NF-kB, Fas-сверхрегуляция, фрагментация ДНК), который может быть предотвращен антагонистами рецепторов интерлейкина-1 [1075]. Кроме того, продукция ИЛ-ф β-клетками отмечается только у пациентов с сахарным диабетом типа II, но не в нормальных островках [1075]. Было показано, что стимулированное ИЛ-1 образование церамида может приводить к активации JNK и фактора, активирующего транскрипцию, которые являются необходимыми, хотя и недостаточными, для индукции NO-синтазы в β-клетках [1743]. Позднее была выяснена роль церамида в цитокин-индуцированной гибели β-клеток [1080] и показано значение активации ERK и MAP-киназы р38 [975, 1296] через стимулированные цитокинами церамиды в экспрессии NO-синтазы и повышении продукции NO· и опосредуемого образованием пероксинитрита апоптоза β-клеток.
Первые экспериментальные работы Robertson et al. показали, что обработка клеточной культуры β-клеток в течение нескольких месяцев высокими концентрациями глюкозы сопровождается значительным снижением экспрессии гена инсулина и активности панкреато-дуоденального гомеобоксного фактора-1 (PDX-1) — основного регулятора синтеза инсулина [1243]. Также было обнаружено, что уменьшение связывания ДНК с другим важным фактором транскрипции, активатором RIPE-3b1 (в настоящее время идентифицированным как MafA), является причиной развития гипергликемии и инсулиновой недостаточности у диабетических крыс [722].
В целом, если в нормальных условиях синтез мРНК инсулина индуцируется в результате связывания с промотором кодирующего его гена факторов транскрипции PDX-1 (три сайта связывания), MafA, β2 и E2A, то в условиях хронического воздействия высоких концентраций глюкозы вследствие нарушения экспрессии PDX-1 (посттранскрипционный дефект) и MafA (посттрансляционный дефект) происходит существенное снижение экспрессии гена инсулина на уровне мРНК, что приводит к уменьшению запасов инсулина и его глюко- зо-стимулированной секреции [1417, 1418] (рис. 18).Вместе с тем при лечении животных троглитазоном, аминогуанидином, флори- зином и N-ацетилцистеином отмечалось повышение ДНК-активности и экспрессии генов PDX-1, MafA и инсулина наряду с улучшением гликемического контроля и снижением выраженности окислительного стресса [721, 722, 1418, 1601]. Поскольку данные препараты обладают антиоксидантной активностью, было сделано заключение, что угнетение экспрессии гена инсулина в результате глюкотоксичности, вызванной гипергликемией, по крайней мере отчасти обусловлено хроническим окислительным стрессом.
Как отмечалось выше, островки Лангерганса содержат относительно малые количества C^Zn-СОД и Mn-СОД, каталазы и ГПО. В 1982 г. Malaisse et al. [1081] выявили, что β-клетки крыс-альбиносов чувствительны к перекисям и активность ГПО в них снижена. Приблизительно в это же время Grankvist et al. [681] обнаружили, что применение СОД предотвращает развитие аллоксанового диабета у крыс. Эти и многие другие исследования доказывают, что низкий уровень островковых антиоксидантных ферментов делает β-клетки наиболее уязвимыми к окислительному стрессу по сравнению с тканями, где уровень антиоксидантной защиты выше.
Может ли глюкоза индуцировать развитие окислительного стресса в островках Лангерганса? Концепция аутоокисления глюкозы, способствующего избыточной генерации АКМ и приводящего к возникновению диабета, была предложена в 1987 г. Вольфом и Дином [1781], хотя в то время его взаимосвязь с нарушением функций β-клеток не была установлена.
На сегодняшний день известно, что в условиях гипергликемии и подавления активности ферментов гликолиза глюкоза окисляется по альтернативным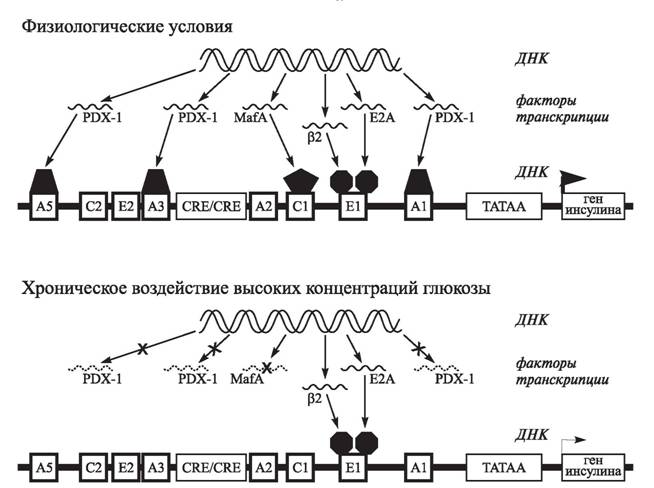
Рис. 18. Нарушение экспрессии гена инсулина в результате глюкотоксичности [1418]
путям, включая полиоловый, гексозаминовый и путь неферментативного гликирования (см. ниже) [1214]. В ходе этих процессов, а также при окислительном фосфорилировании в митохондриях генерируются АКМ [1424], в том числе О2, гидроперекиси, NO·, ОН-радикал. Tanaka et al. показали, что при инкубации островков и клеток HIT-T15 с высокими концентрациями глюкозы возрастает внутриклеточный уровень пероксидов, еще более увеличиваясь при введении в среду бутионинсульфоксимина (ингибитора γ-глутамилцистеинлигазы, ключевого фермента синтеза глутатиона), и это повышение можно предотвратить коинкубацией с манногептулозой, что свидетельствует о том, что именно метаболизм глюкозы является причиной увеличения образования пероксидов в островках Лангерганса [1602]. Было показано также, что свободные радикалы, генерируемые в условиях гипергликемии, способствуют экспрессии NF-κΒ, повышению активности ERK и p38 изоформы MAPK, в результате чего происходит ингибирование се- рин-треониновых фосфатаз, приводящее к пролонгации MAPK-сигнала [1066, 1531].
Влияние липидов на функцию β-клеток, подобно эффектам глюкозы, может быть неоднозначным. Повышение содержания СЖК нарушает секрецию инсулина как in vitro, так и in vivo, предполагается, что это обусловлено аккумуляцией длинноцепочечных эфиров ацетилкоэнзима А в цитоплазме [1130]. Было показано, что длительная экспозиция СЖК в β-клетках ингибирует синтез инсулина на уровне мРНК, а также, отчасти, и стимулированную глюкозой секрецию инсулина [1846]. Вместе с тем существуют и противоположные данные — о позитивном эффекте СЖК на инсулиновую секрецию, в частности, на чувствительность β-клеток к малым концентрациям глюкозы, которая возрастает после продолжительного воздействия высоких концентраций СЖК [350, 411]. Секреторный ответ β-клеток в этих условиях повышался в ответ на низкие и незначительно менялся в ответ на высокие концентрации глюкозы, однако одновременное снижение содержания мРНК и белка инсулина в клетке свидетельствует о том, что механизмы синтеза инсулина более чувствительны к избытку СЖК, чем секреторные механизмы [1827]. В то же время в других экспериментах продолжительная обработка культуры β-клеток СЖК приводила к падению митохондриального мембранного потенциала и открытию АТФ-зависимых К+-каналов, селективно нарушая стимулированную глюкозой секрецию инсулина [968].
Индуцированный жирными кислотами апоптоз, определяемый двукратным повреждением ДНК, проявлялся в значительном повышении уровня церамида в островках предиабетических крыс линии fa/fa при инкубации их в присутствии эфиров Н3-серина и Н3-пальмитина [1505, 1507]. Церамиды могут стимулировать апоптотические процессы за счет активации NF-kB [349], который повышает экспрессию индуцибельной NO- синтазы. В результате усиления генерации NO· возрастает и продукция пероксинитрита — мощного прооксиданта, который является важным проапоптотическим фактором [1031]. По сравнению с нормальными островками, в островках крыс линии fa/fa регистрировалось значительное повышение уровня мРНК индуцибельной NO-синтазы и образования NO· при инкубации с 1 мМ СЖК. Вместе с тем ингибиторы фермента — никотинамид и аминогуанидин — снижали продукцию NO· в этих экспериментах in vitro [1847] и предупреждали развитие диабета и апоптоз in vivo [1506].
Не исключается также и возможность повреждающего эффекта накопления триглицеридов в β-клетках, в результате чего нарушается стимулированная глюкозой β-клеточ- ная пролиферация и секреция инсулина, активируется опосредуемый церамидами JNK/ SAPK-путь апоптоза и повышается продукция NO· [1130, 1668]. Вместе с тем не существует единого мнения относительно механизмов повреждающего действия сверхнакопления липидов на β-клетку. Есть основания сомневаться, что триглицериды ответственны за 15-кратное повышение в инсулинсекретирующих клетках повреждения ДНК, являющегося индексом апоптоза [1505]. Более приемлемо, что триглицериды — лишь отражение неокислительного обмена жирных кислот и источник СЖК в β-клетке.
Поскольку при сахарном диабете типа II отмечается повышение уровня и гликемии, и СЖК, возможно, что именно это сочетание и приводит к максимальному повреждению инсулярного аппарата. Как было показано исследованиями Jasquement et al. [808], длительная инкубация изолированных панкреатических островков или культуры клеток HIT-T15 с высокими концентрациями СЖК и глюкозы снижала как экспрессию инсулиновой мРНК, так и активацию секреции инсулина. Установлено, что липотоксичность β-клеток усиливает глюкозотоксичность и проявляется только в условиях гипергликемии [365, 1335].
Локальным проявлением окислительного стресса в области поджелудочной железы является снижение синтеза инсулина и повреждение β-клеток. Однако гипергликемия, которая наблюдается при диабете как типа I, так и типа II, является причиной развития системных реакций на уровне целого организма.