J ОКИСЛИТЕЛЬНЫМ СТРЕСС. ПАТОЛОГИЧЕСКИЕ СОСТОЯНИЯ И ЗАБОЛЕВАНИЯ L
секретируют около 2 мг инсулина в сутки. Продуцирующие инсулин β-клетки составляют 65 % всей клеточной популяции островков; α-клетки, синтезирующие глюкагон, — 30 %; δ-клетки нарабатывают соматостатин, и на них приходится менее 10 % всех клеток; РР-клетки (0,5 %) служат источником панкреатического полипептида.
Клеточный состав изменяется с возрастом: так, если у новорожденных количество α- и β-клеток одинаково, то в дальнейшем последних становится в 2—3 раза больше. В каждом островке в норме находится около 10 макрофагов [488]; по периферии располагаются вокругос- тровковая капиллярная сеть и нервные сплетения, регулирующие синтез гормонов.При повышении содержания глюкозы в крови она транспортируется в клетки, в том числе в β-клетки островков Лангерганса, в результате усиления гликолиза образуется пируват, который транспортируется в митохондрии и вступает в цикл трикарбоно- вых кислот (рис. 15). Активация последнего сопровождается увеличением содержания в митохондриях NADH и FADH2 и, соответственно, ускорением переноса электронов в дыхательной цепи и синтеза АТФ. Возрастание соотношения АТФ/АДФ приводит к закрытию АТФ-чувствительных калиевых каналов, деполяризации цитоплазматической мембраны и открытию потенциал-управляемых кальциевых каналов, в результате повышения внутриклеточной концентрации ионов Са2+ происходит выброс инсулина из внутриклеточных депо (см. рис. 15).
Первым этапом в инициации биологического эффекта действия инсулина является связывание гормона со специфическими инсулиновыми рецепторами. Инсулиновый рецептор — трансмембранный гликопротеин, относящийся к семейству рецепторов с тирозинкиназной активностью. Он состоит из двух внеклеточных α-субъединиц и двух трансмембранных β-субъединиц, связанных между собой дисульфидными мостиками. Инсулиновый рецептор экспрессируется в виде двух изоформ, различающихся между собой по отсутствию (изоформа А) или наличию (изоформа В) экзона 11, состоящего из 12 аминокислотных остатков [1667]; изоформа А обладает более высоким сродством
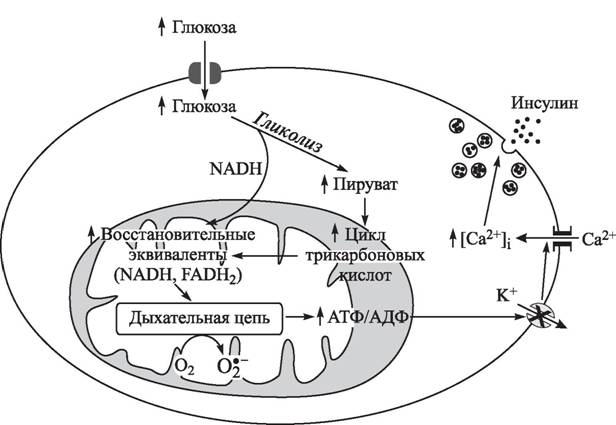
Рис.
15. Глюкозо-стимулированная секреция инсулина в β-клетках поджелудочной железы и генерация АКМ в митохондриях [374]к инсулину. При инсулинрезистентности и сопутствующей ей гиперинсулинемии изоформа А инсулинового рецептора в большей степени подвергается негативной регуляции, и в плазматической мембране изоформа В экспрессируется в большей степени. Обнаружено повышение числа рецепторных изоформ В в скелетной мускулатуре больных диабетом типа II [879], что может быть вторичной причиной инсулинрезистентности и гиперинсулинемии.
После взаимодействия инсулина с α-субъединицей инсулинового рецептора возрастает его тирозинкиназная активность, вследствие чего происходит последовательное аутофосфорилирование нескольких тирозиновых остатков, локализующихся в цитоплазматической части β-субъединицы, с последующим фосфорилированием других внутриклеточных субстратов [863]. Нарушения в процессе аутофосфорилирования рецепторов могут приводить к изменениям в проведении инсулинового сигнала в клетку и к инсулинрезистентности тканей [1218]. Механизмы снижения тирозинкиназной активности инсулинового рецептора, выявляемые при сахарном диабете типа II, остаются неясными. Очевидно, что это обусловлено вторичными метаболическими нарушениями, поскольку мутации гена инсулинового рецептора идентифицированы лишь при редких генетических синдромах с выраженной инсулинрезистентностью [839].
Активация тирозинкиназы инсулинового рецептора необходима для фосфорилирования не только его β-субъединицы, но и других клеточных субстратов, непосредственно взаимодействующих с рецептором. Первый из них был описан в 1985 г. White et al. [1753]. Этот внутриклеточный белок с молекулярной массой 180 кДа был назван инсулинрецепторным субстратом 1 (ИРС-1). ИРС-1, а также последовательно выделенные ИРС-2, -3, -4, -5, обозначаемые сегодня как семейство инсулинрецепторных субстратов, выполняют функцию взаимосвязи между инсулиновыми рецепторами и другими клеточными субстратами, активируя в результате три основных пути передачи инсулинового сигнала: опосредуемый фосфатидилинозитол-3-киназой (PI3K), Cbl/CAP/TC10 и MAPK [1448].
Белки ИРС содержат несколько различных участков: фосфотирозинсвязывающий домен, узнающий аспарагин-пролин-фосфотирозин в юкстамембранном участке инсулинового рецептора, гомологичный домен, который может связываться с мембранными фосфолипидами, и SH2-домен, связывающий фосфорилированные тирозиновые участки ИРС, с последующей активацией различных сигнальных молекул [981, 1585]. Sffi-домены содержат от 50 до 100 аминокислотных остатков и действуют как высокоаффинные фосфотирозинсвязывающие места, которые были найдены в большинстве внутриклеточных сигнальных молекул [1448]. Связывание фосфотирозина с Sffi-доменом приводит к формированию сигнального комплекса [1297].Ключевой сигнальный комплекс формируется между ИРС и регуляторной субъединицей (p85) PI3K. Связывание ИРС с Sffi-доменами субъединицы p85 приводит к активации катализирующей субъединицы фермента, р110, которая фосфорилирует фосфатидилинозитол с последовательным образованием фосфатидилинозитол-3-фос- фата, затем фосфатидилинозитол-3,4-бифосфата и, наконец, фосфатидилинозитол- 3,4,5-трифосфата. Фосфатидилинозитол-3-фосфат действует как внутриклеточный мессенджер, активируя фосфатидилинозитолзависимые киназы и транслокацию глюкозного транспортера GLUT4 в клеточную мембрану, обеспечивая тем самым поступление глюкозы в клетку [1635]. Фосфатидилинозитол-3-фосфат повышает активность каскада протеинкиназ, стимулируя киназу пируватдегидрогеназы [244], которая активирует два класса серин-треониновых киназ: протеинкиназу В и атипичную протеинкиназу С, существующую в двух изоформах (ξ и λ) [982]. Протеинкиназа В фосфори- лирует и регулирует функцию многих клеточных белков, участвующих в процессах клеточного обмена, пролиферации и апоптоза. Так, фермент повышает синтез гликогена путем инактивации киназы гликогенсинтазы-3, которая ингибирует гликогенсинтазу и активирует протеинфосфатазу-1, приводя к активации факторов транскрипции, участвующих в экспрессии гена синтазы жирных кислот, за счет чего стимулируется липогенез [449, 1208].
Семейство PKC также участвует в целом ряде эффектов действия инсулина [982]. В различных тканях разные изоформы PKC транслоцируются в клеточную мембрану из цитозоля в ответ на инсулиновую стимуляцию. Атипичные PKC участвуют в транспорте глюкозы и синтезе белка [926, 1553]. Известно также, что протеинкиназы С могут активировать MAPK и ядерный фактор транскрипции NF-kB, приводя к повышению экспрессии генов и синтезу белка [886].Другой основной путь передачи инсулинового сигнала — MAPK — играет важную роль в регуляции клеточной пролиферации, дифференцировки и апоптозе. Идентифицировано четыре группы MAPK: внеклеточная сигнал-регулируемая протеинкиназа (ERK), c-Jun N-терминальная киназа (JNK), p38-киназа и ER^/большая MAPK-1. Если стимуляторами ERK в основном являются митогенные и ростовые факторы, в том числе инсулин, то киназы р38 и JNK активируются многими средовыми и стрессовыми стимулами (ультрафиолет, ионизирующая радиация и пр.), что приводит к апоптозу. Активация ERK происходит после фосфорилирования ИРС и/или SH-доменов через активацию проонкогенного белка Ras, стимулирующего МАР-киназу с последовательным фосфорилированием и активацией ERK [1576]. Активированная ERK может транслоцироваться в ядро клетки, где катализирует фосфорилирование факторов транскрипции, индуцируя программу клеточной пролиферации и дифференцировки [359].
Сахарный диабет типа II характеризуется двумя основными поломками: нарушением секреции инсулина и снижением чувствительности к нему, или инсулинрезистентностью [531, 1341, 1388]. Инсулинрезистентность, как правило, предшествует развитию сахарного диабета в течение многих лет и, как показано в многочисленных исследованиях [1388, 1389, 1390], чрезвычайно распространена в популяции, выявляясь по меньшей мере у 25 % лиц, не страдающих диабетом типа II. На сегодняшний день получены убедительные данные о генетической детерминированности инсулинрезистентности [247, 531, 624], также свидетельствующие о том, что инсулинрезистентность является главным фактором риска для развития сахарного диабета типа II.
На ранних стадиях инсулинрезистентность компенсируется за счет гиперинсули- немии, в результате чего поддерживается нормальная толерантность к углеводам, однако гиперинсулинемия усугубляет прогрессирование инсулинрезистентности за счет феномена негативной регуляции («down regulation») [531], что требует повышения секреции инсулина. Нарастание этих взаимоусугубляющих нарушений может приводить к нарушению толерантности к углеводам, проявляющей себя в постпрандиальной гипергликемии. По современным представлениям, сахарный диабет типа II манифестирует в ситуации, когда секреторные возможности β-клеток оказываются несостоятельными для преодоления барьера инсулинрезистентности [1388]. Основная роль в этой несостоятельности инсулярного аппарата отводится генетическим нарушениям [624], однако и сама по себе постпрандиальная гипергликемия способна оказывать влияние на снижение секреторных возможностей инсулярного аппарата за счет индукции окислительного стресса. Нарушение утилизации глюкозы периферическими тканями в условиях инсулинрезистентности приводит к активизации липолиза и повышению уровня свободных жирных кислот (СЖК) в плазме, используемых в качестве альтернативного энергосубстрата. Увеличение содержания СЖК и их β-окисление также сопровождается повышением продукции АКМ, усиливающих свободнорадикальные процессы [411, 1794], что приводит к усугублению инсулинрезистентности и снижению секреторных возможностей инсулярного аппарата. Вместе с тем основной причиной инвалидизации и летальности при сахарном диабете типа II является прогрессирование сосудистых осложнений, поражающих как микро-, так и макрососудистое русло. Последние два десятилетия ознаменовались большим количеством исследований, изучавших природу развития этих сосудистых повреждений и приведших к вполне однозначному выводу о ключевой роли гипергликемии в развитии макро- и микроангиопатий [375, 531, 1627].
Механизм индуцированного гипергликемией повреждения тканей, включая макро- и микрососудистое русло, инсулинрезистентность и нарушения в секреции инсулина на современном этапе связывают с окислительным стрессом [375, 1190, 1215]. Его природа при диабете типа II и вопрос о том, развивается ли окислительный стресс на ранних стадиях диабета, приводя к развитию осложнений, или это лишь проявление общего процесса повреждения тканей, отражающее наличие осложнений, имеет принципиальное значение для стратегии лечения: показано ли назначение антиоксидантной терапии для предупреждения тканевого повреждения или достаточно адекватного гли- кемического контроля для профилактики осложнений.
Еще по теме J ОКИСЛИТЕЛЬНЫМ СТРЕСС. ПАТОЛОГИЧЕСКИЕ СОСТОЯНИЯ И ЗАБОЛЕВАНИЯ L:
- Меньщикова Е. Б.. Окислительный стресс: Патологические состояния и заболевания / Е. Б. Меньщикова, Н. К. Зенков, В. З. Ланкин, И. А. Бондарь, В. А. Труфакин.— Новосибирск,2008. - 284 с., 2008
- Механизмы развития окислительного стресса при диабете типа II
- Окислительный стресс и диабетические ангиопатии
- Окислительный стресс и инсулинрезистентность
- Окислительный стресси дисфункция β-клеток при сахарном диабете типа II
- 7.3.1. Автоматизированные системы для распознавания патологических состояний методами вычислительной диагностики
- Психология стресса и функциональных состояний работника
- Психология травматического стресса. Методологические различия при определении стресса и травматического стресса
- Острые заболевания и обострения заболеваний печени, сопровождающиеся болевым приступом
- Окислительная модификация ЛНП и атеросклероз
- Экспериментальные модели окислительного поражения легких у животных
- 42. Стресс и фрустрация
- 6. Патологическая анатомия в России
- Системное изменение активности свободнорадикальных окислительных процессов
- Б. Электронные влияния на окислительно-восстановительные (редокс) потенциалы.
- 7.1. Равновесное состояние изолированной системы как состояние с максимальной энтропией