Ферментативный катализ
Исходным источником энергии для жизни на Земле является солнечный свет, но он никогда не утилизируется непосредственно без предварительной трансформации энергии квантов света в энергию химических связей.
Непосредственным источником энергии для всех типов жизненной активности является химические реакции, идущие с освобождением энергии. Можно сказать поэтому, что важнейшими элементами живой материи являются не просто машины, но химические машины.Белковые катализаторы, т. е. ферменты, ускоряют практически каждый важный внутриклеточный процесс.
Здесь стоит отметить, что проблема функционирования ферментов неотделима от общей проблемы трансформации энергии в биологических системах.
Что такое катализатор? Согласно учебникам физической химии, катализатор — это вещество, влияющее на скорость химических превращений других веществ и не меняющееся при этом. В биохимии принято называть начальные вещества химического превращения субстратами, а конечные — продуктами. Сказанное выше означает, что после каждого акта превращения субстратов в продукт фермент возвращается в свое исходное состояние. Исходное и конечное состояния молекул фермента не изменяется, в то время как концентрация молекул субстрата и продукта меняется с каждым оборотом фермента.
Существует огромная литература, касающаяся как экспериментальных, так и теоретических аспектов ферментативного катализа. Я затрону здесь только те из них, которые имеют прямое отношение к главной проблеме этой главы.
Л. Михаалис сформулировал классический подход к описанию процесса трансформации субстрат-продукт под влиянием фермента. В соответствии с этим подходом, молекулы субстрата и фермента прежде всего образуют так называемый комплекс Михаэлиса, в котором превращение субстрата в продукт идет легче, чем в случае свободного субстрата. Схема ферментативной реакции может быть представлена следующим образом: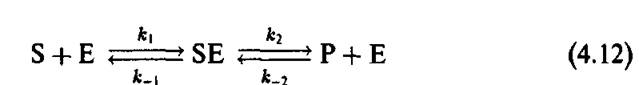
Здесь к2 и к_2 — константы скорости соответствующих ре
акций.
Анализ этой схемы в рамках формальной химической кинетики приводит к знаменитому уравнению Михаэлиса—Ментен:
Здесь Уо — начальная скорость реакции, т. е. скорость образования продукта (d[P]/d£), [Р] — концентрация продукта, [S] — концентрация субстрата, Утах — максимальная начальная скорость реакции (при S —► оо), Км — константа Михаэлиса:

Согласно (4.13) константа Михаэлиса равна концентрации субстрата, при которой скорость реакции равна половине максимальной:
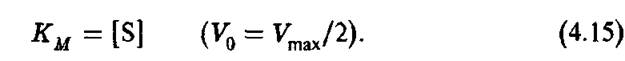
Зависимость скорости реакции от концентрации субстрата описывается гиперболической кривой (рис. 4.5). Согласно Михаэлису Км является главной характеристикой ферментативной реакции для данного субстрата при фиксированных значениях температуры, pH и т.д. Широко используемая схема Михаэлиса ферментативного катализа
основана на классической химической кинетике, развитой в конце XIX века для химических реакций низкомолекулярных соединений в газовой фазе или разбавленных растворах. Все последующие теории ферментативного катализа используют, в конечном счете, ту же основу [1,31]. Однако, как было сказано выше, химические процессы с участием макро молекулярных соединений (белков) протекают при возбуждении особых механических степеней свободы. В литературе было независимо предложено два
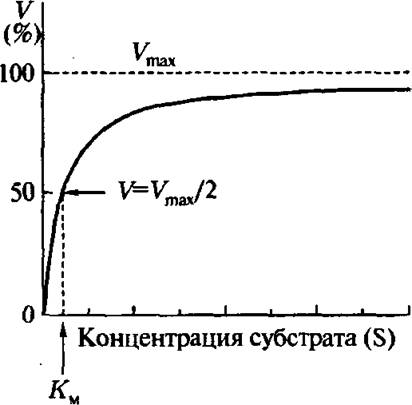
Рис. 4.5. Зависимость скорости ферментативной реакции от концентрации субстрата (по Михаэлису)
очень близких подхода к ферментативному катализу и процессам внутриклеточной трансформации энергии, учитывающие механические аспекты функционирования ферментов.
Конечно, истинная история этих подходов включает множество работ и ученых (почти полную библиографию можно найти в уже цитированных книгах), но согласованный анализ ситуации связан с двумя группами исследований: первая — из Московского института химической физики и вторая — из Лондонского университета.Я начну с релаксационной концепции ферментативного катализа. Первое указание на отклонение каталитического акта ферментативной реакции от классической термодинамики и классической кинетики было, по-видимому, высказано в 1971 году [34]. Было показано, что применение основных постулатов Аррениуса и Эйринга к большинству ферментативных процессов может привести к бессмысленным значениям активационных параметров. Функционирование фермента больше похоже на работу механической конструкции, чем на обычную каталитическую химическую реакцию. Феноменологическая самосогласованная релаксационная теория ферментативного катализа была предложена в 1972 году [35,36]. Принципиальная идея релаксационной концепции заключается не просто в том, что конфирмационная
релаксация субстрат-ферментного комплекса связана с изменением каталитической активности фермента.
Присоединение субстрата к активному центру фермента инициирует конформационную релаксацию, действующую как движущая сила, которая толкает химическую систему (молекулы субстрата, связанные с активным центром фермента) вдоль координаты реакции.
Любое локальное химическое изменение молекулы белка (присоединение субстрата или ингибитора к активному центру, редокс изменения металла в простетической группе, ионизация кислотной или основной группы и т.д.) приводит к появлению конформационно неравновесного состояния. Быстрая колебательная релаксация активного центра и его ближайшего окружения происходит немедленно после локального возмущения, в то время как структура основной белковой глобулы остается практически неизменной. Молекула становится неравновесной. Новое, кинетически доступное состояние фермент-субстрат- ного комплекса соответствует конформационно измененной структуре с продуктом, связанным с активным центром.
Превращение субстрата в продукт реализуется в ходе конформационной релаксации фермент- субстратного комплекса к новому состоянию равновесия. Этот переход протекает чрезвычайно медленно по сравнению с масштабом времени колебательной релаксации. В некоторых случаях этот процесс занимает несколько сотен миллисекунд или даже несколько секунд [37]).Схема ферментативной реакции может быть представлена теперь следующим образом:
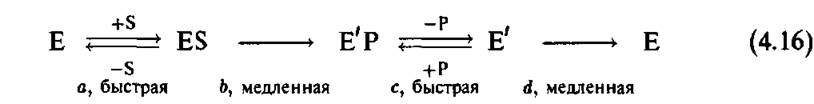
Здесь Е' означает конформационно измененную молекулу фермента.
Эта предельно упрощенная схема включает два типа стадий:
1. Быстрые обратимые стадии связывания и отцепления (стадии а и с).
2. Стадии медленной необратимой релаксации (стадии b и d).
Химические превращения S --------------- ► Р — это один из элементарных
актов схемы (4.16). На рис. 4.6 изображена разветвленная схема с набором траекторий, ведущих от ES к Е'Р (стадия b в уравнении 4.16).
Для любой сложной глобулы фермента имеется множество возможных путей и возможных промежуточных неравновесных состояний фер- мент-субстратного комплекса. На рис. 4.7 показан профиль потенциальной энергии при движении системы вдоль одного из возможных
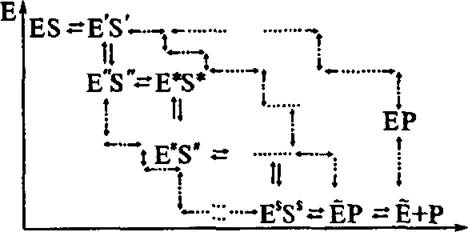
Рис. 4.6. Разветвленная схема ферментативной реакции
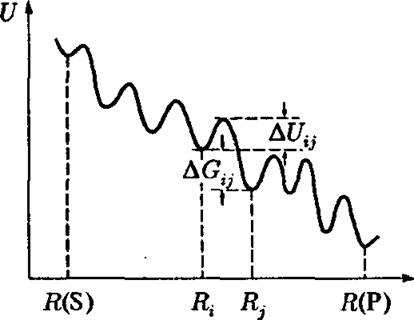
Рис. 4.7. Профиль потенциальной энергии вдоль одного из возможных путей на схеме 4.6
путей от S к Р. Конечно, вероятности реализации различных путей отличаются друг от друга. Каждый элементарный акт на рис. 4.6 начинается с образования нового неравновесного состояния системы и его последующей релаксации к следующему состоянию после преодоления соответствующего барьера. Среди огромного числа возможных траекторий только немногие будут непосредственно участвовать во всем процессе релаксации фермент-субстратного комплекса.
Мы можем сказать, что релаксационный процесс в определенной степени детерминирован. В течение этого процесса релаксировавшая часть белковой глобулы постепенно увеличивается и «механическое движение» релаксирующей молекулы фермента происходит под действием силы, возникающей между релаксировавшей и не релаксировавшей частями макромолекулярного комплекса.Возможность микроскопического описания этого релаксационного процесса, начиная с первых принципов, практически исключена.
Хотя каждый элементарный акт обратим, весь процесс типа Ь (так же как и d) практически необратим. Это вынуждает нас рассматривать эволюцию системы от ES до Е'Р как механическое движение вдоль выделенной степени свободы, которое закончится после достижения равновесного состояния.
Итак, согласно релаксационной концепции, скорость химического превращения субстрата в продукт определяется, как правило, скоростью конформационной релаксации. Температурная зависимость скорости обусловлена не изменениями числа молекул субстрата, способных преодолевать активационный барьер, а изменениями конструкции фермент-субстратного комплекса, которые влияют на путь и, следовательно, на скорость конформационной релаксации. Из приведенного анализа можно сделать еще один важный вывод: прямой и обратный пути реакции, катализируемые ферментом, могут существенно отличаться. Это означает, что «релаксационная схема» может быть реализована только вне термодинамического равновесия системы субстрат-продукт. Другими словами, если фермент «работает» как механическая машина, то механизмы реакции вблизи термодинамического равновесия и вдали от него должны различаться.
Соответствующие экспериментальные данные и их детальный теоретический анализ, так же как и соответствующие ссылки, можно найти в цитируемых выше монографиях.
4.6.
Еще по теме Ферментативный катализ:
- Ферментативная деструкция.
- Ферментативные антиоксиданты
- Постановка проблемы
- Структура, функциональные и некоторые физикохимические свойства ацетилхолинэстеразы
- ПЕРОКСИДНОЕ ОКИСЛЕНИЕ ЛИПИДОВ КАК ОДИН ИЗ КЛЮЧЕВЫХ МЕХАНИЗМОВ МОДИФИКАЦИИ СТРУКТУРНО-ФУНКЦИОНАЛЬНОГО СОСТОЯНИЯ БИОМЕМБРАН
- J ОКИСЛИТЕЛЬНЫМ СТРЕСС. ПАТОЛОГИЧЕСКИЕ СОСТОЯНИЯ И ЗАБОЛЕВАНИЯ L
- Миелопероксидаза
- Ксантиноксидаза
- Защита миокарда от свободнорадикального поражения
- ОПРЕДЕЛЕНИЕ ФУНКЦИОНАЛЬНОЙ АКТИВНОСТИ НЕКОТОРЫХ АНТИОКСИДАНТНЫХ ФЕРМЕНТОВ КРОВИ[6]