4.4. Основы термодинамики и кинетики химических реакций
Начнем с элементарных актов в простой химической реакции

Здесь и к2, соответственно, константы скорости при единичных концентрациях реагентов прямой и обратной реакции.
Константа Равновесия оеакции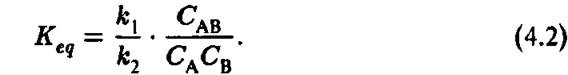
Здесь — концентрация і-го компонента реакции в условиях химического равновесия. На рис. 4.3 представлена традиционная схема изменений энергии во время химической реакции. Ось абсцисс Является так называемой «координатой реакции» Д, т. е. обобщенные относительные положения всех атомов реагирующей молекулы во время перехода от левого состояния к правому состоянию (рис. 4.3). По оси ординат отложена полная энергия химической системы. Параметры на схеме рис. 4.3 и обычно используются для
описания химических реакций в рамках классической химической

Рис. 4.3. Изменения энергии системы в ходе химической реакции
термодинамики и кинетики. Параметр ZLff° — это стандартное (т. е. при единичных концентрациях реагентов) изменение энтальпии системы. Его значение равно «теплоте реакций», т. е. полному изменению энергии системы, вызванному химическим превращением. Кинетические параметры Е? и — энергии активации прямой и обратной реакций соответственно. Согласно квантовой механике все эти параметры должны отсчитываться от дискретных энергетических уравнений, а не от дна энергетической ямы. В обеих ямах для начальных и конечных продуктов низшими являются т. н. нулевые энергетические уровни, соответствующие температуре Т = О К. Для адекватного термодинамического описания химической реакции обычно необходимо знать стандартное изменение энтропии, т.
е. разницу энтропии равновесных начального и конечного состояния химической системы. Изменения энтропии в ходе химической реакции могут зависеть от пути реакции, так что было бы ошибочным откладывать непрерывные изменения энтропии на схеме, аналогичной приведенной на рис. 4.3. Непрерывная линия изменений энергии на рис. 4.3 означает, что мы молчаливо предполагаем, что при каждом значении координаты реакции R энергия в химической системе успевает достичь равновесия в соответствии с непрерывно изменяющимся состоянием системы. Это необязательно верно.Главное уравнение химической термодинамики, связывающее  с константой равновесия К'
с константой равновесия К'
су
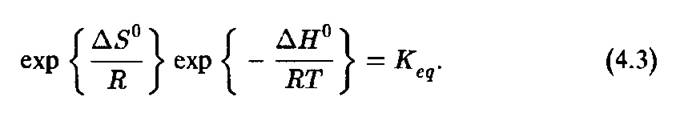
Это уравнение эквивалентно следующему:

Здесь ДС?° представляет стандартное изменение свободной энергии Гиббса в результате химической реакции. Для «идеальных систем» величины К в уравнениях 4.3, 4.2 эквивалентны. Уравнение 4.2 позволяет в принципе определять экспериментально константу равновесия, измеряя равновесные концентрации реагентов.
Измерение температурной зависимости константы равновесия К позволяет определить значение с помощью уравнения Вант- Гоффа.
с помощью уравнения Вант- Гоффа.
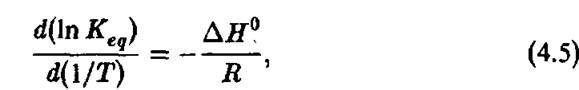
і а затем рассчитывать Д5° с помощью уравнения 4.3. Этот подход Вант-Гоффа используется в подавляющем большинстве литературных данных по термодинамическим параметрам биохимических реакций. Откладывая IgK, экспериментально измеренный при разных температурах, против 1/Т, можно рассчитать значения Д5°, Этот широко используемый подход молчаливо предполагает, что Д5° не зависят от температуры или удовлетворяют следующему уравнению
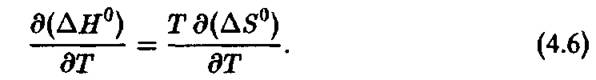
В> рамках общепринятого термодинамического подхода изменение ДЯ° и Д5° с температурой вызывается только изменением распределения реагирующих молекул по Больцмановским уравнениям, в то время как формы потенциальных ям (рис.
4.3) остаются независимыми от температуры. Мы увидим далее, что это условие может не выполняться для многих биохимических реакций, катализируемых ферментами.Существует простой метод проверки правильности определения ДН° с помощью уравнения Вант-Гоффа. Тепловой эффект химической реакции можно измерить непосредственно калориметрическим методом. Однако, не существует методов прямого измерения активационных параметров, определяющих скорость химической реакции.
Согласно знаменитому уравнению Аррениуса константа скорости химической реакции равна

Здесь Еа — энергия активации (см. рис. 4.3). Предэкспоненци- альный («частотный») множитель А равен числу столкновений реагирующих молекул за единицу времени. Множитель А предполагается независимым от температуры. В этом случае

Поэтому Еа можно рассчитать, откладывая экспериментально измеренные значения In А: против 1/Т. Согласно простой теории столкновения химической кинетики, акт химического превращения может произойти, только если суммарная кинетическая энергия сталкивающихся молекул превосходит активационный барьер Еа. Экспоненциальный множитель в (4.7) определяет долю таких столкновений, которые могут привести к акту химической реакции. Отметим, что (4.7) предполагает выполнение Больцмановского равновесного распределения энергий молекул в реакционной смеси.
Эйринг предложил более сложный подход в своей теории активированного комплекса (см. например, [33]). Обобщенная координата реакций R на рис. 4.3 символизирует путь между исходной и конечной конфигурациями молекул, которые нужно пройти, чтобы преодолеть низший возможный потенциальный барьер. Максимальное значение этого барьера (точка «а. с.» на рис. 4.3) является седловой точкой: она представляет максимум потенциальной энергии при движении вдоль координаты реакции и минимум потенциальной энергии вдоль других направлений.
Статистический расчет, используемый при таком подходе, внутренне противоречив. При расчете предполагается, что между начальным состоянием и «активированным комплексом» существует термодинамическое равновесие, которое сохраняется в течение всего времени химического превращения. Мы увидим, что во многих случаях, особенно для ферментативных реакций, это не верно. Ниже показана упрощенная форма уравнения Эйринга для константыскорости
I
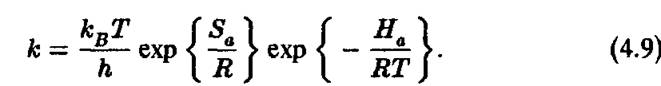
Здесь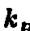 j — константа Больцмана, h — константа Планка, Sa
j — константа Больцмана, h — константа Планка, Sa
и ЯЛ, соответственно, энтропия и энтальпия активации. Энтальпия активации Яв в (4.9.) играет роль энергии активации в уравнении Аррениуса (4.7). Между этими величинами нет различия (см., напри-
мер, [1]).
Состояние активированного комплекса не является реальной молекулой и поэтому не может быть непосредственно фиксировано и «измерено». Его время жизни носит характер бесконечно малой величины. Скорость реакции, таким образом, это число молекул, проходящих через состояние активированного комплекса за единицу времени. Теория Эйринга предполагает, что это происходит бесконечно быстро. Это означает, что скорость реакции определяется числом химических превращений в единицу времени, а не длительностью отдельного элементарного акта.
Расчет химической кинетики в рамках подходов Аррениуса и Эйринга основан на постулатах классической равновесной термодинамики. Предполагается, что химическая система проходит через ряд равновесных состояний. Принцип микроскопической обратимости действует на всем пути от исходных до конечных продуктов. Отсюда следует, что пути прямой и обратной реакции совпадают. Как было подчеркнуто выше, однако, активационные параметры Еа и Sa нельзя измерить непосредственно. Необходимо поэтому оценить применимость общепринятых подходов физической химии, предложенных много лет назад для описания поведения низкомолекулярных соединений в газовой фазе и в разбавленных растворах для биохимических процессов.
Для сложных молекул (например, белков) изменение температуры может привести не только к изменению Больцмановского распределения молекул по уровням энергии, но и к изменениям структуры белка. Такие конформационные изменения могут изменить форму потенциальных ям, характеризующих процесс (см. рис. 4.3). Это означает, что формулы (4.5) и (4.8) могут оказаться неверными и использование уравнения Вант-Гоффа в термодинамике так же, как и Аррениуса и Эйринга в кинетике, могут привести к ошибочным результатам. Так,
Часть II. Основные решаемые проблемы 6*.
например, небольшие изменения конфигурации белка с температурой могут вызывать значительные изменения активационного барьера, рассчитанного по Аррениусу или Эйрингу, для химического превращения субстрата, связанного с ферментом, без заметных изменений истинной энергии активации Sa. В узком температурном интервале, обычном при исследовании ферментативных реакций, любая слабая температурная зависимость энергии активации Еа может быть аппроксимирована линейным законом: Еа = В + ЬТ, где В и Ь — константы.
Подставляя Еа в уравнение (4.7) или (4.9), получаем:
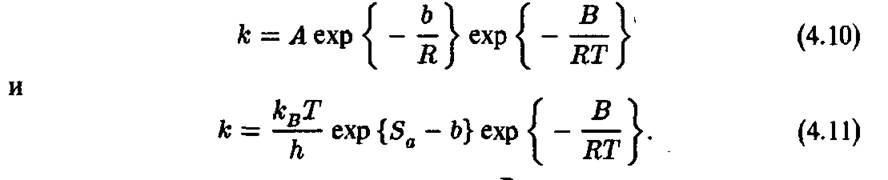
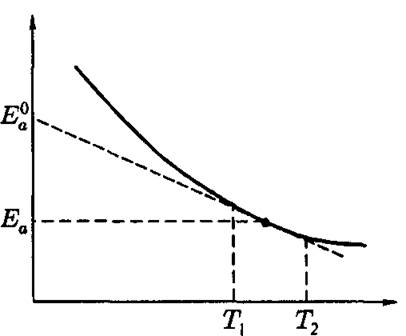
Рис. 4.4. Истинная и кажущаяся энергии активации
В этом случае экспериментальные данные будут формально удовлетворять уравнению Аррениуса. Однако мы будем измерять не истинные активационные параметры Еа и Sa, но эффективные величины В = Еа - ЬТ и Sa - b, соответственно, которые могут существенно отличаться от истинных значений. Ясно, что величина В представляет собой экстраполяцию к О К, полученную из реальной температурной зависимости Еа (рис. 4.4). Даже если Е очень слабо зависит от температуры
в исследуемом температурном интервале г, - 12, разница между ис-
Т, - Г.
тинным и кажущимся значениями активационных параметров может быть велика.
Сказанное справедливо и для уравнения Вант-Гоффа (4.5).
В классической химической кинетике скорость реакции опреде
ляется как число мгновенных актов в единицу времени. Если, однако, конформационная релаксация белков является частью элементарного химического акта, последний не может считаться мгновенным.
Белки, как и другие макромолекулярные компоненты живой материи, представляют собой не только статистические, но и механические системы. Это значит, что во многих случаях, когда мы имеем дело с промежуточными (неравновесными) состояниями макромолекул, а не просто с квазиравновесными, исходными или конечными состояниями системы, использование свободной энергии становится бессмысленным. Механика оперирует только полной энергией и ее производной по расстоянию, т.е. силой. Такой «механический» подход к ферментативному катализу и биоэнергетическим процессам должен поэтому включать понятие силы.
4.5.