4.0.4. Лекарственные вещества, разрушающие ДНК
Доксорубицин (4.27), называвшийся ранее адриамицином, антибиотик ряда антрациклинов, широко используется для лечения карциномы и саркомы. В присутствии доксорубицина у природной ДНК повышается температура плавления и вязкость, уменьшается плавучая плотность и константа седиментации.
Это указывает на его принадлежность к интеркаляторам. Об этом же свидетельствует смещение видимого спектра и спектра флуоресценции доксирубицина в комплексе с ДНК, а также легкость его восстановления. В результате изучения серии его производных было установлено, что их карциностатическая активность падает при уменьшении способности соединения и интеркаля- ции, а также при снижении основности. Был сделан вывод о том, что высокоосновная аминогруппа в углеводной части молекулы доксорубицина образует ионную связь с фосфатной группой ДНК (так же как это было показано кристаллографически для дауномицина), а плоский антрахиноновый цикл вклинивается между слоями оснований [di Marco, Arcamone, 1975]. Доксорубицин ингибирует ДНК-зависимую РНК- и ДНК-полимеразу (последнюю — на 50% в концентрации 7,4 мг/мл).Как считают некоторые авторы, кроме интеркалирующих свойств, доксорубицин обладает способностью разрушать ДНК путем активации супероксиддисмутазы [Oberley, Buettner, 1979], однако повреждающее действие доксорубицина на ДНК пока еще не доказано, и в качестве противоположной точки зрения можно привести предположение о воздействии его на поверхность клетки [Tritton, Уее, 1982; Israel, Potti, 1982]. О клиническом использовании доксорубицина см. Blum, Carter (1974) и Gottlieb, Hill (1974).
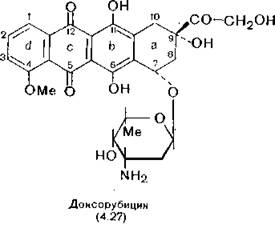
Доксорубицин выделяют из Streptomyces peucetiues, при этом выделяется и даунорубицин, отличающийся наличием в молекуле группы —СОСНз вместо —СОСН2ОН. Даунорубицин используется в основном при лейкозах [Arcamone et al., 1969].
Ярко выраженная способность дауномицина к интеркаляции изучена наиболее полно. Кристаллографически установлено, что углеводная часть молекулы располагается в малой бороздке двойной спирали, метоксигруппа оказывается в большей бороз- ке, а гидроксильная группа в положении 9 антрациклиновой системы образует водородную связь с ближайшим гуанином [Quigley et al., 1980]. Это подтверждается данными ЯМР-спектров, из которых видно, что парой оснований перекрываются кольца С и В [Jain, Kozlowski, Rice, 1981].Основной недостаток доксорубицина — его кардиотоксичность. В поисках менее токсичного противоопухолевого препарата исследовали ряд его аналогов, некоторые из них были получены синтетическим путем (4/-тетрагидропиранилдоксорубицин) или выделены из родственных видов растений (карминомицин или аклациномицин A) [Carter, Sacurai, Umezawa, 1981]. О действии доксорубицина см. обзоры Schwartz (1983); Neidle, Sanderson (1983) и El Khadem (1982).
Блеомицины — семейство антибиотиков, выделенных в Японии из Streptomyces verticillus [Umezawa, 1973]. Фармакопейный блеомицин — это смесь блеомицина А2 (4.28) и блеомици- иа В2, отличающегося от А2 тем, что триметиленсульфониевая группа (на формуле снизу справа) замещена тетраметиленгуа- нидиновой (—NH) (CH2)4NHC( = NH)NH2 [Oppenheimer, Rodrigues, Hecht, 1979]. При добавлении различных аминов в ферментационный бульон было получено около 400 других блеоми- цинов. Из них наиболее перспективный, по-видимому, пепломи- цин, так как он более устойчив к метаболическому гидролизу и менее токсичен по отношению к легочной ткани. Блеомицин широко применяют в клинической практике, он быстро купирует солидные формы рака и входит в небольшое число препаратов, не повреждающих костный мозг. Побочные эффекты блеомици- на (фиброз легких и некоторые поражения кожи) отличаются от таковых других противоопухолевых препаратов, применяемых при солидных опухолях (например, доксорубицина и цисплатина).
Поэтому при парентеральном введении блеомицина в комплексе с одним из этих препаратов противоопухолевый эффект аддитивно возрастает, а токсическое действие на здоровье клетки уменьшается до минимума [Carter, Blum, 1976; Carter et al.,, 1976].He воздействуя на РНК, блеомицин разрушает одноцепочечные ДНК в опухоли и препятствует образованию пар [Nagai et al., 1969].
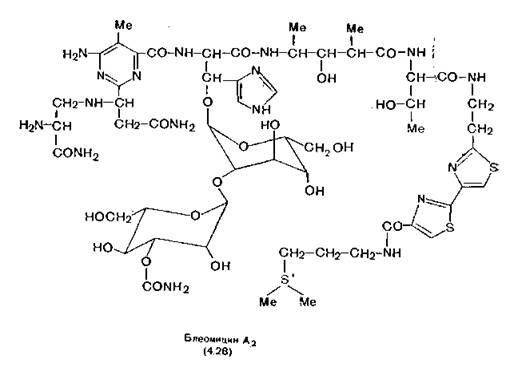
Структура молекулы блеомицина (ОММ. более 1500) на первый взгляд довольно сложна. Однако при ее рассмотрении становится понятным, что активность блеомицина определяется наличием в молекуле двух областей: интеркалирующей (справа на формуле 4.28) и хелатирующе-окисляющей (слева). Так же как и молекулы аминоакридинов (разд. 2.2),.молекула блеомицина за счет своей высокоосновной группы (сульфониевой или гуанидиниевой) связывается с фосфатным анионом в ДНК- При сближении двух молекул плоский битиазолиловый фрагмент блеомицина проскальзывает между парами оснований ДНК- Интеркаляция приводит к тесному соединению молекул, что обеспечивает активность второй части молекулы блеомицина [Takita et al., 1978; Chien, Grollman, Horowitz, 1977].
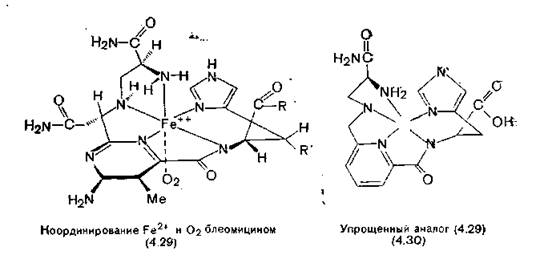
В организме блеомицин связывается с ионом железа. Пять, атомов азота блеомицина образуют с ним координационные связи (4.29), а шестая связь железа захватывает молекулу кислорода, превращая ее в гидрокси-анион-радикад ('О-)[8] [Sausville, Peisach, Horowitz, 1978]. К сожалению, ни сам блеомицин, шт его соль с железом пока не удается получить в кристаллическокг состоянии, приемлемом для рентгеноструктурного исследования, хотя из блеомицина легко получают хорошо кристаллизующиеся* медные соли. Рентгеноструктурный анализ показывает располо* жение шести тяжелых атомов из левой части молекулы рядом с атомом меди (4.28).
Активированный комплекс (4.29) окисляет атом углерода в положении 4'-дезоксирибозильного остатка в ДНК раковых клеток [Grollman, Takeshita, 1980].
В результате происходит окислительное расщепление связи между атомами С-3 и С-4 дезоксирибозы и образуются четыре 3-замещенных пропеналя, например тимин-3-пропеналь (4.31) [Giloni et al., 1971].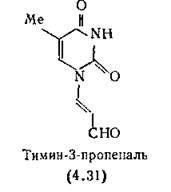
Синтезированный группой Umesawa аналог (4.30) обладает;., подобно блеомицину, способностью связывать ион железа и ак-
•тивировать кислород, но не разрушает ДНК, так как в его мо- .лекуле нет интеркалирующих групп. Однако молекулы, в которых железосвязывающая группа ковалентно связана с интерка- лирующим остатком, разрушают цепи ДНК так же, как и блеомицин. В качестве примеров следует упомянуть гемин, присоединенный к 9-аминоакридину [Lown, Joshua, 1982], и ЭДТА (разд. 11.6), присоединенную к аминофенантридину [Hertzberg, iDervan, 1982].
Известно, что тимин-3-пропеналь (4.31) проявляет отчетливые цитотоксические свойства; в связи с этим до сих пор неясно, ‘Обусловлен ли противоопухолевый эффект блеомицина образованием при распаде ДНК опухолей 3-замещенных пропеналей оснований.
Флеомицины, выделенные из тех же Streptomyces, отличаются от блеомицинов только наличием двух дополнительных атомов водорода в тиазольном цикле (при этом гетероцикл остается плоским). При окислении флеомицина образуются соответствующие блеомицины [Umezaw, 1973]. Противоопухолевые -свойства этих соединений еще только начинают изучать.
Высокоактивные «усилители» действия флеомицина и блеомицина, азотсодержащие гетероциклы, были открыты Brown и ‘Grigg (1982). Наиболее активным, видимо, является 2-(5',7'-ди- метил-5-триазоло[1,5-с]пиримидин-2-илтио) ацетамид [Allen et al., 1983]. Действие «усилителей», вероятно, отчасти обусловлено тем, что они расплетают двойную спираль ДНК, делая ее бо- .лее доступной для антибиотиков [Grigg, Edward, Brown, 1971]. Возможно также, что они блокируют энергетический обмен в митохондриях до завершения распада ДНК.
Добавляя в среду к бактерии-продуценту различные амины, можно получать более эффективные аналоги блеомицина.
Соединения, в которых сульфониевая или гуанидиниевая группы заменены менее основной аминогруппой, значительно сильнее повреждают опухоли, чем легкие. Одно из таких производных, пепломицин, отличающийся от (4.28) только тем, что группа —NH(CH2)3S+Me2 заменена на —NH(CH2)3NHCH(Me)Ph, в настоящее время проходит интенсивные клинические испытания в Японии [Carter, Sacurai, Umezawa, 1981]. В США тщательно изучают блеомицин-ВАРР, имеющий группу —NH(CH2)3NH(CH2)3NH(CH2)3NH, и таллисомицин—флеоми- /цин с таким же заместителем, имеющий еще один остаток аминоуглевода.Зиностатин (неокарциностатин) и ауромицин — два других антибиотика, разрывающих ДНК- Однако не нашли они широкого клинического применения.
Молекулярные аспекты противоопухолевой терапии на примере алкилирующих агентов рассмотрены в разд. 13.4, а также в работе Neidle, Waring (1983).
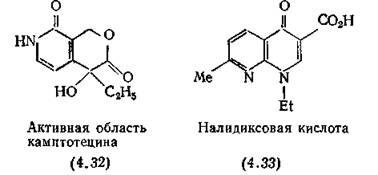
Камптотецин, алкалоид из тибетского кустарника Camptot- heca acuminata, разрезает ДНК таким образом, что она может =158
быть сшита клеточными лигазами [Horowitz, Brayton, 1970]. Его молекула содержит пять конденсированных циклов, активность определяется наличием пиридинового лактона (4.32). Камптотецин используют главным образом в биохимических исследованиях, так как он ингибирует биосинтез рибосомной и матричной ДНК, но не влияет на синтез митохондриальной РНК. [Abelson, Penman, 1972]. Противолейкозные свойства камптоте- цина описаны Wall, Wani (1977).
Кофеин увеличивает скорость спонтанного разрушения ДНК. Е. соН и является для нее сильным мутагеном [Grigg, 1970],. однако миллионы людей ежедневно принимают его в больших, количествах. Согласно правилам регистрации лекарственных: веществ, существующим в развитых странах, этого побочного» эффекта кофеина достаточно, чтобы запретить потребление чая и кофе. Однако следует иметь в виду, что большинство мутировавших клеток нежизнеспособно, а небольшой процент мутаций» в действительности полезен.