АТЕРОСКЛЕРОЗ И ОКИСЛИТЕЛЬНАЯ МОДИФИКАЦИЯ ЛИПОПРОТЕИНОВ НИЗКОЙ ПЛОТНОСТИ
Атеросклероз (от греч. αθαρη — «кашица» и σκληροσ — «твердый») — хроническое заболевание артерий, сопровождающееся разрастанием множественных плотных узловатых утолщений стенки артерий (бляшек), суживающих ее просвет и способствующих тромбообразованию.
Это — широко распространенное заболевание: так, около половины всех людей, живущих в высокоцивилизованных странах, умирают от осложнений атеросклеротического процесса в сосудах различных органов (инфарктов, инсультов и др.). Только в США и Европе (включая Россию) в ХХ в. от последствий атеросклероза умерло более 360 млн человек, что много больше числа жертв всех войн, происшедших в этом столетии [186]. Атеросклероз сосудов считается главной причиной роста смертности от сердечно-сосудистых заболеваний, которая в 1900 г. составляла 1 %, а к 2000 г. выросла до 50—60 %.Развивается атеросклероз в результате сложных структурных изменений в интиме (внутреннем слое) и в медии (мышечном слое) артерий, которые характеризуются на первом этапе накоплением липидов и холестерина в виде внеклеточных липидных частиц и массивных отложений этерифицированного холестерина в составе так называемых «пенистых» клеток, а на более поздней стадии — разрастанием соединительной ткани и увеличением холестерино-фиброзных и кальциевых отложений. Первые работы по изучению состава атеросклеротических бляшек выполнены в начале прошлого столетия. В результате многочисленных патоморфологических, гистохимических и биохимических исследований было показано, что основная масса липидов и эфиров холестерина в бляшке происходит из циркулирующих в крови липопротеинов низкой плотности (ЛНП) [589]. Главным маркером на начальной этапе атеросклеротического процесса является появление в интиме сосудов пенистых клеток, имеющих в цитоплазме массивные липидные включения, в большинстве своем представленные эфирами холестерина [759].
Пенистые клетки были идентифицированы как макрофаги моноци- тарного происхождения и пролиферирующие гладкомышечные клетки [1816], при этом основная масса пенистых клеток имеет моноцитарное происхождение [235].В 1912 г. русским ученым Н. Н. Аничковым было показано, что кормление кроликов холестерином приводит к поражению сосудов, во многом идентичным наблюдаемому при атеросклерозе у людей. Последующие клинические исследования показали, что повышенный уровень холестерина в крови у людей является важным фактором риска развития атеросклероза. Так, среднее содержание холестерина в плазме больных (6,31 ммоль/л) значительно выше, чем у здоровых людей (4,99 ммоль/л), при этом увеличивается холестерин, преимущественно входящий в состав ЛНП (4,40 ммоль/л; в норме — 3,14 ммоль/л); одновременно наблюдается гипоальфахолестеринемия — снижение уровня холестерина в липопротеинах высокой плотности (1,06 ммоль/л по сравнению с 1,42 ммоль/л у здоровых людей), обладающих способностью выводить холестерин из клеток периферических тканей в печень, где происходит его катаболизм [61, 175]. Поэтому атеросклероз можно отнести к патологиям, вызванным нарушением холестеринового обмена (по меткому определению Н. Н. Аничкова, «без холестерина нет атеросклероза»); отношение общего холестерина в сыворотке к α-холестерину является показателем атерогенности [21]. Липидная, или холестериновая, теория атеросклероза подтверждается многими эпидемиологическими исследованиями; кроме того, воздействия, снижающие общий холестерин и холестерин в составе ЛНП, замедляют или останавливают развитие патологического процесса.
Повышенное содержание холестерина в крови — фактор риска, но не достаточное условие атеросклеротических изменений в сосудах. Как отмечают многие исследователи, другим необходимым условием атерогенеза является повреждение стенки сосуда, что приводит к секвестрации и накоплению в интиме фагоцитирующих клеток с последующим их перерождением в «пенистые» клетки. Такое рассмотрение участия клеточных элементов в патогенезе позволяет отнести атеросклероз к своеобразной форме хронического воспаления, в качестве начальной фазы которого выступает адгезия к эндотелию гранулоцитов, вызывающих его повреждение [1002]; а ключевым элементом формирования атеросклеротической бляшки являются моноциты-макрофаги, трансформирующиеся в пенистые клетки в результате захвата модифицированных ЛНП [1180].
Правомочность рассмотрения атеросклероза с такой точки зрения подтверждается наличием признаков хронического воспаления различной интенсивности в стенках атеросклеротически пораженной аорты [1425], при этом интенсивность пролиферации клеток и экспрессия молекул клеточной адгезии VCAM-1, ICAM-1 в этих участках коррелирует с выраженностью воспалительной реакции [38, 1728]. Считается, что обострение течения такой хронической формы воспаления является причиной повреждения стенки сосуда и образования сгустка крови в области развития бляшки, что часто служит причиной летальных исходов.Толчком в изучении роли окисленных ЛНП в атерогенезе послужило открытие в макрофагах специальных «скэвинджер»-рецепторов (рецепторов-«мусорщиков») для модифицированных ЛНП [373]. Первоначально они были описаны лауреатами Нобелевской премии Гольдштейном и Брауном как рецепторы к ацетилированным ЛНП [669]. Однако дальнейшие исследования показали их высокую аффинность для ацетоацетилированных, карбамилированных, сукцинилированных, обработанных малоновым диальдегидом и окислено модифицированных ЛНП, определенных полири- бонуклеотидов (политимин), полисахаров (декстрансульфат, каррагинан), хемокинов, апоптотических телец, некоторых фосфолипидов и бактериальных липополисахаридов [935, 1184]. Общим признаком всех лигандов для скэвинджер-рецепторов является наличие полианионных комплексов (рецептор-«липучка»). В отличие от В/Е-рецепторов для нативных ЛНП скэвинджер-рецепторы связывают только модифицированные ЛНП, и их экспрессия не регулируется внутриклеточным содержанием холестерина [373]. На сегодня в клетках млекопитающих описано 8 классов разных по структуре скэвинджер-ре- цепторов, для каждого из которых существуют свои предпочтительные лиганды (рис. 5). Больше всего скэвинджер-рецепторов выявляется в моноцитах/макрофагах (если не рассматривать клетки печени, где содержание рецепторов на порядок выше); в эндоте- лиоцитах их мало, а в гладкомышечных клетках они обнаруживаются после обработки форболовыми эфирами или продуктами тромбоцитарного происхождения [1331].
Основным скэвинджер-рецептором для окисленных ЛНП в макрофагах является CD36. В человеческих моноцитах ИЛ-4 и М-КСФ повышали, а ЛПС и дексаметазон снижали экспрессию гена CD36 [1813]. В культурах мышиных макрофагальных клеток RAW 264,7 экспрессия CD36 увеличивалась в ответ действие окисленных липопротеинов и ИЛ-4, при этом экспрессия регулировалась протеинкиназой С (PKC) и ядерным фактором транскрипции PPAR-γ [599]. В макрофагах дефицитных по PPAR-γ наблюдается низкая экспрессия мРНК и белка CD36, что свидетельствует о регуляции базального уровня CD36 ядерным макрофагальным фактором транскрипции — PPAR-γ [445]. Окисленные ЛНП, 4-гидрокси-2-ноненаль и электрофильный агент диэтилмалеат индуцировали экспрессию CD36 посредством активации ARE, одновременно повышался синтез ряда антиоксидантных ферментов, что делало клетки более устойчивыми к токсическому действию окисленных ЛНП и АКМ [796].
Фактически можно считать, что начало свободнорадикальной теории атерогенеза было положено в начале 1980-х гг., когда было показано, что инкубация ЛНП с эндоте- лиоцитами приводит к их окислительной модификации и усилению поглощения макрофагами [748, 1565]. Захват окисленных ЛНП макрофагами происходит через скэвинджер-рецепторы и приводит к накоплению в них эфиров холестерина с последующим формированием пенистых клеток, во многом схожих с клетками атеросклеротических бляшек [373, 514, 1816]. Одновременно было показано, что окисление ЛНП делает их цитотоксичными в отношении разных типов клеточных культур [751]. Тот факт, что
формирование пенистых клеток происходит при инкубации макрофагов только с окисленными или модифицированными («атерогенные ЛНП»), но не с нативными ЛНП, послужил основой концепции, согласно которой начальным этапом атерогенеза является возникновение окисленных ЛНП, цитотоксичных для эндотелиоцитов и усиливающих адгезию нейтрофилов, что вызывает повреждение эндотелия; захват окисленных ЛНП макрофагами приводит к образованию пенистых клеток, их накоплению в интиме сосудов и в последующем — к формированию атеросклеротической бляшки (рис.
6). Это позволило А. Н. Климову утверждать, что «без атерогенных липопротеинов не будет атеросклероза» [93]. При этом, несмотря на наличие различных механизмов модификации ЛНП in vitro, окислительная модификация считается наиболее вероятной и наиболее доказанной in vivo в организмах животных и человека.Таким образом, радикальные окислительные реакции с участием АКМ являются атерогенным фактором, так как вызывают модификацию липопротеинов (преимущественно ЛНП) плазмы крови; кроме того, АКМ и продукты их реакций, в том числе и окисленные липопротеины, оказывают цитотоксическое и деструктивное действие на клетки и ткани [417, 745], что может лежать в основе повреждения сосудов [113, 115]. Множество экспериментальных, клинических и эпидемиологических исследований позволяют отнести атеросклероз к классическим свободнорадикальным патологиям [1, 41]:
• при атеросклерозе наблюдается усиление свободнорадикального окисления и накопление продуктов ПОЛ (изопростанов F2, гидроперекисей холестерина, ТБК-РП) как в области атеросклеротической бляшки, так и в крови; как правило, выявляется местное и общее снижение уровня антиоксидантной защиты [85, 118, 125, 1332];
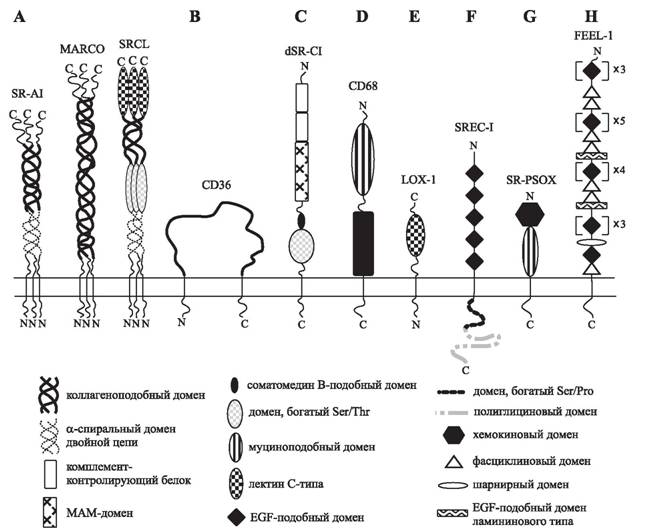
Рис. 5. Структура различных классов скэвинджер-рецепторов (A-H) эукариот [1184]
1 39 г
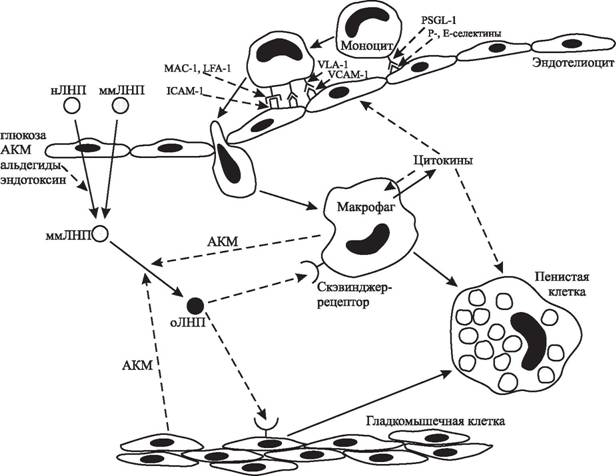
Рис. 6. Механизмы возникновения атеросклеротического поражения сосудов. нЛНП — нативные ЛНП, ммЛНП — минимально модифицированные ЛНП, оЛНП — окисленные ЛНП
• применение антиоксидантов предотвращало развитие атеросклеротических повреждений сосудов у экспериментальных животных и снижало риск развития сердечно-сосудистых заболеваний у людей [1191];
• рентгеновское облучение мышей инициировало формирование атеросклеротических бляшек [1646].
При этом из приведенной на рис. 7 схемы видно, что свободнорадикальная гипотеза этиологии атеросклероза не только не противоречит двум другим общепризнанным гипотезам, объясняющим возникновение атеросклеротических бляшек липидной инфильтрацией стенки сосуда или повреждением сосудистого эндотелия, но и существенно дополняет их.
Приведенные в этом разделе данные дают также основание полагать, что повышенное содержание липопероксидов в плазме крови (гиперлипо- пероксидемия) является одним из биохимических маркеров атеросклероза и наряду с гипоантиоксидантемией может рассматриваться в качестве дополнительного фактора риска этого заболевания. При этом очевидно, что гипоантиоксидантемия и гиперли- попероксидемия не являются специфичными признаками атеросклероза, но важный вклад свободнорадикальных процессов в этиологию и патогенез заболеваний сердечнососудистой системы на основании имеющихся в настоящее время экспериментальных и клинических данных представляется несомненным.Наряду с увеличением концентрации продуктов ПОЛ в сыворотке, и особенно в ЛНП, имеются данные, что у больных атеросклерозом снижается собственная биохемилюминесценция сыворотки, которая отражает образование радикалов RО· [69]. Это подтверждается экспериментальными исследованиями на кроликах, содержавшихся на рационе с высоким содержанием холестерина: в первые 50 дней у них в 3 раза повышалось спонтанное свечение сыворотки, а в дальнейшем, при развитии атеросклеротического поражения сосудов, интенсивность хемилюминесценции снижалась [68]. Из этих данных делается вывод, что при атеросклерозе процессы ПОЛ в сыворотке тормозятся, и выдвигается гипотеза, согласно которой атеросклероз представляет собой адаптационное состояние, защищающее организм от поражения сосудов и мембран клеток продуктами ПОЛ; защита осуществляется трудноокисляемыми липидными соединениями. Выявлены также другие механизмы компенсации токсического воздействия ПОЛ на организм. Так, некоторые гидроперекиси холестерина проявляют антипролиферативный эффект и ингибируют образование жировых полосок у животных, содержащихся на атерогенной диете [1032, 1633]. Антиокислительным и антиатерогенным действием обладают липопротеины высокой плотности (1,125—1,210 г/мл) [92]; кроме того, будучи окислены, они ингибируют эндогенный синтез холестерина в клетках [655].
Выявление атерогенной роли окисленных ЛНП породило интереснейшую проблему: что же лежит в основе и является причиной окислительной модификации ЛНП в организме in vivo? Свободнорадикальные реакции с участием АКМ, несомненно, служат основой окисления ЛНП [171, 584, 589, 703], вместе с тем конкретные механизмы не ясны, возможно участие в этом процессе продуктов ПОЛ, таких как МДА или 4- гидроксиноненаль [837, 1264]. У курильщиков в 1,5 раза повышено содержание МДА в сыворотке, при этом у молодых людей курение в 5 раз увеличивает риск развития ишемической болезни сердца [1299, 1579]. Проведенные в последние годы исследования клеточных ферментативных механизмов генерации АКМ выявили участие данных
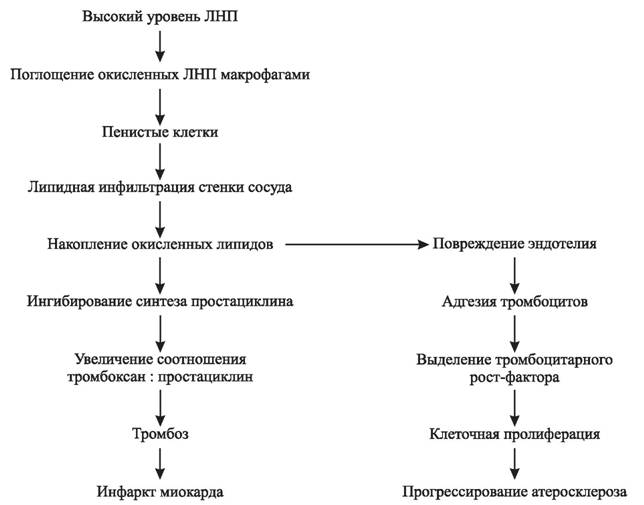
Рис. 7. Упрощенная схема, объединяющая липидно-инфильтрационную гипотезу атеросклероза, свободнорадикальную гипотезу и гипотезу, объясняющую возникновение атеросклеротических бляшек повреждением эндотелия
ферментных систем в окислении ЛНП [770, 822, 941]. Гликозилирование белковой компоненты ЛНП [93, 283] или изменение их липидного слоя в результате действия различных фосфолипаз [281, 572] и холестериноксидазы [282] также может приводить к появлению атерогенных ЛНП и накоплению холестерина и эфиров холестерина в макрофагах. Таким образом, проблема модификации ЛНП и формирования пенистых клеток в процессе атерогенеза далека от своего окончательного разрешения. Вместе с тем множество данных указывает на то, что реакции с участием АКМ являются необходимым атрибутом процесса атерогенеза.