Фармакокинетика
Так называется область науки, изучающая кинетику процессов всасывания, распределения, метаболизма и выведения лекарственных веществ. Самые ранние исследования по фармакокинетике общих (ингаляционных) анестетиков [Wildmark, 1920; Dominguez, 1933] неприменимы для изучения других соединений.
Изучая действие инсулина, Teorell (1937) вывел основные уравнения фармакокинетики. Он предложил использовать обычные формулы химической кинетики для определения концентрации лекарственного препарата в месте его введения, затем вкрови и тканях и, наконец, после инактивации или элиминации. Применение этих уравнений получило дальнейшее развитие в работах Kriiger-Thiemer (1960) и Nelson (1961). На сегодняшний день фармакокинетику лекарственного вещества считают его важнейшей характеристикой, подлежащей обязательному изучению. Знание фармакокинетики позволяет установить необходимую для больного дозу лекарственного вещества и оптимизировать режим его введения. Контролируя концентрацию лекарственного препарата и его метаболитов на каждой стадии распределения, по данным фармакокинетики можно определить стадию, на которой следует повысить избирательность.
Даже для соединений, близких друг к другу по химическому строению, на разных стадиях распределения (см. рис. 3.2) существуют фармакокинетические различия, во многом определяющие избирательность действия лекарственных веществ. Метаболизм каждого лекарственного препарата в организме описывается суммой констант, значения которых обусловлены особенностями химической структуры этих соединений (для разных видов животных эти константы различны).
Отсутствие у клиницистов данных по фармакокинетике может приводить к двум крайностям: применению недостаточной дозы (не дающей эффекта) и передозировке (приносящей вред больному). Для создания оптимальной схемы лечения необходимо контролировать: а) эффективность действия лекарственного вещества и б) продолжительность его действия.
Время проявления лечебного эффекта зависит от скорости всасывания и распределения препарата, продолжительность действия определяется скоростью метаболизма и выведения, а величина дозы в сочетании с этими параметрами определяют эффективность лекарственного вещества.Данные о распределении вещества, полученные в экспериментах на животных, только приблизительно отражают его истинные фармакокинетические свойства, поэтому необходимо проводить клинические испытания.
Время нарастания концентрации лекарственного вещества в крови при внутривенном и пероральном введении резко отличается (рис. 3.6). Очевидно, что при внутривенном введении в крови сразу создается максимальная концентрация препарата, тогда как при пероральном она нарастает постепенно, достигая более низкого уровня. Скорость уменьшения действия препарата в том и другом случае одинакова.
Скорость прохождения лекарственного вещества через мембрану может быть представлена в дифференциальной форме как dS/dt, где dS — микроскопическое количество вещества, прошедшее через мембрану за очень малый промежуток времени (разд. 3.2). Скорость переноса лекарственного вещества через мембрану прямо пропорциональна его количеству (S) на внешней поверхности мембраны:
—dS/dT=kS,
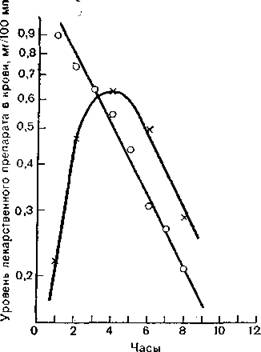
Рис. 3.6. Средний уровень теофиллина в крови человека при внутривенном (кружки) и пероральном (крестики) введении.
где к—константа проницаемости мембраны, So — количество вещества в нулевой момент времени (до всасывания), т. е. доза (разд. 3.2).
Кажущийся объем распределения (Vd) (разд. 3.1) определяется либо для одной камеры организма (обычно, для крови), либо для двух — при переходе лекарственного вещества из крови, например, в ткани. В связи с тем что инфекционный процесс развивается в основном в тканях, а не в крови, распределение лекарственного вещества между кровью и тканями способствует проявлению его химиотерапевтического действия.
Двухфазный характер кривой зависимости концентрации лекарственного вещества от времени (первого порядка) подтверждает распределение его между двумя камерами организма. Константа ki2 характеризует переход агента из камеры 1 в камеру 2, тогда как k2i относится к обратному процессу.Кинетику двухкамерного всасывания изучали Loo, Riegel- man (1968). Сердечный гликозид дигоксин — типичный представитель препаратов, фармакокинетика которых может быть описана двухкамерной моделью. В случае барбитуратов периферической камерой является липоидная ткань.
В отличие от константы проницаемости, связанной с вводимой дозой, все остальные константы, независимо от того, относятся они к переходу лекарственного вещества из одной камеры в другую или к выведению его из организма, определяются его
концентрацией. Если внести небольшие изменения в приведенное выше уравнение, то оно приобретет вид:
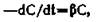
где С — концентрация вещества перед прохождением через мембрану, р — константа выведения или всасывания, а —dC/dt— «скорость исчезновения». Константа выведения обычно берется как сумма констант — метаболической и экскреции. Наряду с наиболее часто встречающимися константами первого порядка существуют константы нулевого порядка, т. е. не зависящие от количества (или концентрации) присутствующего лекарственного вещества [Nelson, O’Reilly, 1960, 1961].
Для того чтобы установить оптимальную схему дозировки, определяют период полураспада лекарственного вещества, представляющий собой время, за Которое половина его количества выводится из кровяного русла (to,5). Зная скорость выведения лекарственного вещества, можно рассчитать to 5 по формуле: t0,5=0,693/0.
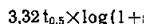
Идеальный интервал доз (т) равен
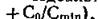
.
........... где Со представляет собой исходную концентрациюв крови, a Cmin — минимальную терапевтическую эффективную концентрацию. Однако рассчитанные по этому уравнению интервалы времени между введениями препарата практически трудно соблюдать (например, 19 ч), поэтому обычно устанавливают более удобный режим приема лекарственных веществ (через 4, 8, 24 или 48 ч). Подставляя эти значения т в следующее уравнение, находят оптимальную дозу (D) [Wagner, 1957]:
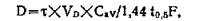
где Cav — средний необходимый уровень в крови, F—абсорбированная фракция (в идеале 1,0), а остальные константы имеют свое прежнее значение. Ниже приведен список наиболее широко применяемых препаратов и периоды их полураспада в организме человека в часах [подробнее см. Gilman, Goodman и Gilman, 1980, р. 1675].
| Алпреиолол | 3 |
| Ампициллин | 1,3 |
| Анаприлин | 4 |
| Апрессин | 2 |
| Аспирин | 0,3 |
| Варфарии | 37 |
| Гентамицин | 2 |
| Дигитоксин | 7 |
| Дигоксин | 42 |
| Изониазид | 2 |
| Цмизин | 13 |
| Индометацин | 2 |
| Клонидни | 9 |
| Левомицетин | 30 |
| Метилдофа | 1,8 |
| Метотрексат | 8 |
| Морфин | 3 |
| ІІреднизолон | 2 |
| Рифамииции | 2 |
| Снбазон | 50 |
| Сульфаметоксозол | 9 |
| Тетрациклин | 10 |
| Теофиллин | 9 |
| Трнметоприм | 11 |
| Тубокурарин | 2 |
| Фенобарбитал[6] | 86 |
| Хинидин | 6 |
| Хлортиазид | 1,5 |
| Цефалексин | 0,9 |
| Циметидин | 2 |
| Эритромицин | 1 |
| Этанол | 0,2 |
Таблица 3.5. Рекомендуемые интервалы между дозами н соотношение первичной дозы (D*) к поддерживающей (D) дли получения устойчивого уровни концентрации в крови
| Средний период полураспада, ч | Интервал времени между дозами, ч | D*/D | |
| Норсульфазол | 3,5 | 4 | 1,8 |
| Сульфанзоксазол | 6,1 | 6 | 2,0 |
| Стрептоцид | 8,8 | 8 | 2,1 |
| Ацетилсульфаизоксазол | 13,1 | 12 | 2,1 |
| Сульфазин | 23,5 | 24 | 3,0 |
| Сульфамеразин | 23,5 | 24 | 3,0 |
| Сульфадиметоксин | 41,0 | 24 | 3,0 |
больших дозах, будет накапливаться в организме, особенно если его to,5 велико.
Данные о режиме дозировки сульфамидов, приведенные в табл. 3.5, были получены следующим образом: лекарственный препарат вводили перорально здоровым добровольцам и через небольшие промежутки времени брали у них пробы крови и мочи для анализа. Таким же образом были определены константы скоростей метаболизма и выделения (рис. 3.7). Сложная константа выделения может быть представлена в виде суммы двух микроскопических констант, характеризующих выведение вещества почечными клубочками и обратное всасывание его из почечных канальцев в кровь.
Исследования противобактериальных сульфаниламидов показали, что их фармакокинетические свойства улучшаются с повышением липофильности: увеличивается всасываемость, а также кажущийся объем распределения, хотя в последнем случае ионизация играет незначительную роль. Скорость выведения в равной степени зависит от липофильности вещества и его способности к ионизации. С повышением липофильности увеличивается связывание вещества с белками (исключение составляют только орто-изомеры, что связано со стерическим влиянием заместителя), но затрудняется их метаболизм (например, ацетилирование) . В противоположность вышесказанному, в экспериментах in vitro противобактериальные свойства сульфаниламидов мало изменяются с изменением липофильности, но значительно зависят от электронных эффектов, особенно от способности молекул к ионизации (разд. 10.5), и усиливаются при введении орто-заместителей [Seydel, 1981]. Эти исследования, равно как и изучение других групп лекарственных веществ, представляют особый интерес с точки зрения проблемы избирательности, так как свидетельствуют о том, что действие лекарственных веществ определяется разнообразными, подчас совершенно неожиданными независимыми параметрами. Отсюда следует, что на характер распределения лекарственных веществ можно воздействовать, даже незначительно изменяя их молеку-
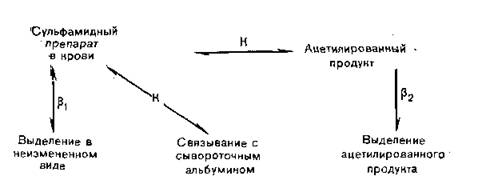
Рис.
3.7. Кинетика метаболизма и выведения сульфамидных препаратов из организма человека.лярную структуру. Таким образом, перед исследователями открываются новые возможности повышения избирательности действия лекарственных препаратов путем регуляции распределения (подробнее см. разд. 9.3).
Использование пролекарств требует определения фармакокинетических констант как для пролекарств, так и для лекарственного вещества [Martin, 1967]. Если скорость выведения предшественника меньше таковой самого лекарственного вещества, то концентрация последнего в крови при введении его в виде пролекарства снижается медленнее, чем при введении самого препарата. Аналогично влияет на уровень содержания лекарственного вещества в крови скорость превращения пролекарства в истинное лекарство. В результате настойчивых поисков может быть создано пролекарство с исключительно удачным сочетанием значений констант скоростей обоих вышеуказанных процессов. Такое лекарственное вещество можно будет вводить через большие промежутки времени, одновременно поддерживая постоянный уровень его концентрации между введениями.
Всасывание твердых лекарственных препаратов при пероральном введении — медленный и экспоненциальный процесс. Скорость всасывания пропорциональна величине поверхности и убывает в ряду: растворы, суспензии, капсулы, прессованные таблетки, таблетки, покрытые оболочкой. Натриевые соли слаборастворимых кислот создают в крови более высокую концентрацию, чем свободные кислоты, так как при взаимодействии этих солей с соляной кислотой желудочного сока соответствующая слабая кислота выделяется в значительно более дисперсной форме, чем та, в которой выпускаются лучшие из имеющихся в продаже препаратов. Кинетика растворения лекарственных соединений рассмотрена в обзоре Wagner (1961).
Биодоступность слаборастворимых препаратов при пероральном введении значительно варьирует из-за разного размера их частиц. Измерение увеличения концентрации лекарственных веществ в плазме выявило разницу между различными формами аспирина, дифенилгидантоина, сердечных гликозидов (особенно дигоксина), тетрациклина, левомицетина и дикумарина.
3.7.2.