Принципы химической кинетики
Итак, теперь можем предсказать распределение температуры во времени и в пространстве. Но каков механизм диссоциации молекулы при нагреве и как зависит скорость такой диссоциации от температуры?
Механизм равновесной тепловой диссоциации.
В условиях термодинамического равновесия распределение энергии по степеням свободы при достаточно высоких температурах оказываетсяблизким к больцмановскому, т.е. число молекул на каждом энергетическом уровне ~ exp(- E/kT), где E - энергия этого состояния. Это означает, что при любой температуре найдется какая-то доля молекул, заселяющих даже довольно высокие колебательные подуровни (рис. 5.2). Другое дело, что при низких температурах эта доля близка к нулю, однако, никогда точно не равна нулю. Следовательно, молекулы, для которых энергия возбуждения выше энергии диссоциации (см. рис. 5.2) будут диссоциировать. Ясно, что при увеличении температуры больцмановское распределение сдвигается «вверх» по рисунку, и количество таких диссоциировавших молекул будет расти.
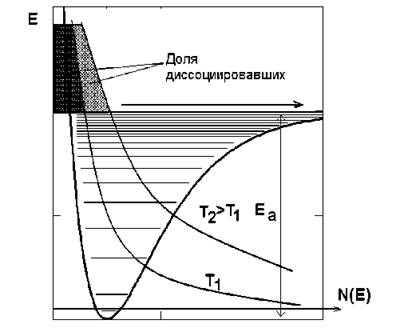
Рис. 5.2. Механизм равновесной диссоциации молекул при различных температурах. Кривые: терм, больцмановское распределение при температуре Т1, больцма- новское распределение при температуре Т2
При достаточно высокой температуре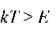 доля таких моле
доля таких моле
кул становится уже порядка единицы. Теперь понятна последовательность разрывов связей при плавном росте однородно распределенной температуры: вначале диссоциируют связи с наименьшей энергией разрыва связи (так называемые энергии активации) Ea, затем с несколько большей и т.д. Этим и будет определяться состав химических продуктов при пиролизе в конденсированных средах.
Реакция диссоциации с точки зрения химии всегда является мономолекулярной. В растворах, однако, возможны столкновения молекул разного сорта, таких, что в результате может образоваться новая химическая связь. Такие реакции называются бимолекулярными (или тримолекулярные). Для каждой реакции характерной величиной является скорость: число актов реакции в единицу времени. Вопросами скорости реакции занимается химическая кинетика.
Элементы химической кинетики. Основные принципы химической кинетики чрезвычайно просты. Целью нашего рассмотрения будет только построение уравнений, описывающих зависимости концентраций веществ от времени при протекании химических процессов [11]. Как увидим, эти уравнения оказываются подобны уравнениям кинетики состояний, полученным в гл. 2.
Химическая кинетика изучает скорость реакции, зависимости скорости от различных факторов, а также пути протекания реакции. Кинетика изучает влияние на скорость химических реакций состояния реагирующих веществ и их концентрации, присутствия посторонних веществ, температуры, воздействия различных излучений. Кинетика занимается изучением механизма химических процессов и разработкой теории таких процессов. Ввиду того, что химические процессы часто комбинируются с процессами растворения, адсорбции и другими физическими процессами, вопросы химической кинетики тесно связаны с вопросами кинетики ряда физических процессов.
Формальная кинетика изучает зависимость скорости реакции (протекающей при постоянной температуре) от различных факторов, а также занимается классификацией химических реакций. В химии считается, что формальная кинетика обычно не объясняет характера наблюдаемых зависимостей и детального механизма протекающих процессов. Процессы изучаются и классифицируются на основе нескольких принципов, принимаемых за аксиомы. К их числу относится закон действующих масс, который позволяет выразить скорость химической реакции с помощью концентраций реагентов. Суть этого закона состоит просто в том, что количества атомов данного сорта всегда сохраняются и поэтому убыль одних молекул вследствие реакции означает прибавление других.
В равновесии, как гласит закон действующих масс, скорости прямой и обратной реакции равны. Количество молекул данного сорта, участвующих в элементарном акте реакции есть стехиометрический коэффициент. Для элементарной реакции
(где а и b - стехиометрические коэффициенты исходных веществ реакции, Р - продукты реакции) по закону действующих масс скорость реакции (число актов реакции в единицу времени) равна 
где сА и сВ - концентрации веществ А и В. Константа k называется константой скорости реакции. В химии концентрации обычно измеряются в молях на литр (моль/л, напомним, что моль - это число молекул, равное числу Авогадро, ~ 6 · 1023), но в физике конденсированных сред, где объем системы почти всегда можно считать неизменным, обычно пользуются концентрациями, выраженными в просто в долях от полного числа молекул в кубическом сантиметре конденсированной среды (т.е. в см-3).
Другим принципом формальной кинетики является положение о том, что в случае сложной реакции, состоящей из нескольких отдельных стадий, эти стадии протекают независимо друг от друга и скорость одной из них никак не влияет на скорость остальных. Как закон действующих масс, так и принцип независимого протекания отдельных стадий не являются абсолютными, а их безусловное использование в формальной кинетике, как считается в химии, указывает на ее ограниченную применимость.
В ходе всякой реакции изменяется количество участвующих в ней веществ. Поэтому изменение количества какого-либо реагента за определенное время может также характеризовать скорость реакции. С другой стороны, при постоянном объеме системы скорость химической реакции численно равна изменению концентрации любого из реагирующих веществ в единицу времени, деленному на соответствующий стехиометрический коэффициент:
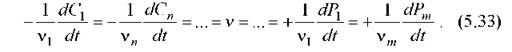
Здесь Ci - концентрации исходных веществ, а Pi - концентрации продуктов реакции.
Принято считать скорость реакции положительной величиной, поэтому скорости изменения концентраций исходных продуктов (которые в реакции убывают) взяты со знаком минус. Понятно, откуда берется стехиометрический коэффициент: если, например, в реакции участвует сразу две молекулы вещества А, то скорость изменения его концентрации будет вдвое выше, чем скорость реакции (определяемая как число актов в единицу времени).Из приведенных выше уравнений видно, что константа скорости численно равна скорости данной реакции в случае равенства единице концентраций всех веществ. Константа скорости зависит от тех же факторов, что и скорость данной реакции, кроме концентрации реагирующих веществ и времени. Если какая-либо реакция протекает при постоянной температуре и при постоянстве других условий, константа скорости этой реакции является определенной величиной и может характеризовать реакцию. В отличие от этого, скорость реакции в качестве ее характеристики непригодна, так как постоянно изменяется в ходе большинства реакций. Поясним еще два часто встречающихся понятия. Молекулярность химической реакции равна числу исходных молекул, принимающих участие в элементарном акте этой реакции. В зависимости от числа таких молекул различают мономолекулярные, бимолекулярные и тримолекулярные реакции. Порядок химической реакции равен сумме показателей степени концентрации реагентов в кинетическом уравнении реакции (т.е. в уравнении для скорости реакции). Порядок и молекулярность реакции, как видим, должны совпадать. Несовпадение порядка реакции, определенного в эксперименте, с ожидаемой молекулярностью обычно говорит о том, что происходят какие-то промежуточные реакции, которые не учтены той моделью, из которой вычислялась молекулярность.
Теперь приведем более или менее обобщенный пример составления уравнений химической кинетики на основе этих принципов. Пусть имеет место следующая реакция (протекающая как в прямом, так и в обратном направлении):
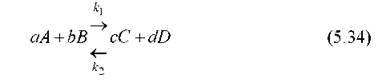
где ki и k2 - константы скорости прямой и обратной реакции.
Тогда, пользуясь изложенным выше, получим уравнения, описывающие изменения концентраций всех веществ во времени: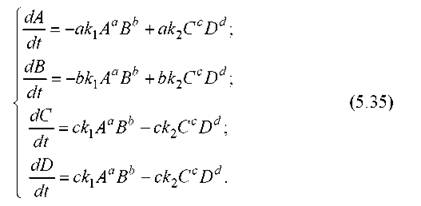
Здесь концентрации обозначены теми же символами, что и сами вещества. Маленькими буквами обозначены их стехиометрические коэффициенты. В правых частях стоят слагаемые, определяющие скорость производства соответствующего вещества со знаком «плюс», скорость его убыли - со знаком «минус».
Стехиометрические множители типа 1/а здесь перенесены в правые части уравнений. Это сделано для удобства по следующей причине. Допустим, одно и то же вещество А участвует сразу в нескольких реакциях. Тогда мы должны добавить в правую часть дифференциального уравнения для dA/dt слагаемые, отвечающие производству или убыли А в каждой такой реакции. Естественно, здесь уже нельзя приравнивать скорости разных реакций, но, по смыслу, скорость производства (убыли) А в другой реакции просто суммируется со скоростью производства (убыли) в данной реакции. Поясним это примером. Пусть, например, одновременно протекают две реакции, в которых участвует одно и то же вещество А 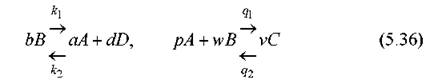
(Здесь p, w, v - стехиометрические коэффициенты второй реакции.) Тогда, по описанным правилам, получим дифференциальное уравнение для A:
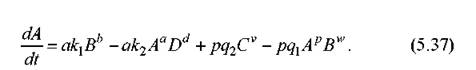
Итак, эти простые правила позволяют построить уравнения химической кинетики в большинстве практических случаев.
Физическое обоснование химической кинетики. Заметим, что при стехиометрических коэффициентах, равных единице, уравнения химической кинетики математически полностью совпадают с уравнениями кинетики населенности состояний, выведенными в гл. 1. Такое совпадение не случайно. Как видели, уравнения кинетики населенностей выводились из первых принципов, точнее, из нестационарного уравнения Шредингера.
Как нетрудно понять, происхождение уравнений химической кинетики, в общем, имеет ту же природу. Так, если реакция мономолекулярна, то это очевидно: мы просто можем считать исходную молекулу одним состоянием квантовой системы, а результирующую молекулу - другим. Ясно, что к этим двум состояниям, в принципе, применим развитый выше формализм кинетики населенностей. Несколько сложнее случай немономолекулярной реакции. В этом случае следует применять развитые ныне в химии представления о т.н. активированном комплексе. Активированный комплекс - комплекс, образующийся при столкновении всех исходных молекул в левой части уравнения реакции. Понятно, что по принципам статистической физики вероятность такого столкновения будет пропорциональна концентрации каждой из участвующих молекул. Если, например, реакция требует участия сразу двух молекул А (т.е. стехиометрический коэффициент равен двум), то вероятность такого столкновения, очевидно, будет пропорциональна квадрату концентрации и т.п. Тем самым, вероятность образования активированного комплекса пропорциональна концентрациям всех молекул, взятым в степенях их стехиометрических коэффициентов. Следующий этап - как и при мономолекулярной реакции - есть просто квантовый переход между возможными состояниями уже одной и той же квантовой системы (активированного комплекса).Таким образом, как видим, ограничения, накладываемые применением химической кинетики, проявляются в основном тогда, когда неизвестен механизм процесса. Если механизм процесса известен, то правильно построенные уравнения химической кинетики обычно дают правильное предсказание поведения системы.
Зависимость скорости реакции от температуры. Из рис. 5.2 ясно, что с повышением температуры скорости даже мономолекулярных химических реакций (диссоциации), как правило, сильно возрастают. То же происходит и в случае немономолекулярных процессов. Существует приближенное правило Вант-Гоффа: при
повышении температуры на 10 °С скорость реакции увеличивается
приблизительно в 2 - 4 раза. В соответствии с этим правилом, изменение температуры на 100 °С приводит к изменению скорости реакции примерно в 310 « 60 тыс. раз. Более точно температурная зависимость скорости химической реакции выражается уравнением Аррениуса [11]:
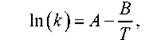
где k - константа скорости данной реакции; Т - абсолютная температура; А и В - эмпирические постоянные. Эту же зависимость часто записывают в форме
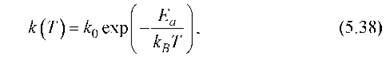
вводя вместо А и В другие постоянные величины: k0 - предэкспоненциальный множитель и Еа - энергию активации. Именно это выражение (5.38) обычно называют формулой Аррениуса (рис. 5.4). Физический смысл k0 и Ea объясняется на основе представлений статистической физики и термодинамики. Пусть изменение энергии системы в описанных выше физических представлениях в ходе протекания химической реакции изменяется так, как представлено на рис. 5.3. Уровень I соответствует исходным веществам, уровень II - продуктам реакции. Из рисунка видно, что в ходе реакции энергия системы сначала возрастает от уровня I до точки К, а затем уменьшается до уровня II. Как говорят, в ходе реакции преодолевается энергетический барьер. Высота этого барьера характеризуется той минимальной дополнительной энергией E1 по сравнению со средней энергией теплового движения молекул, которую нужно сообщить исходным веществам для того, чтобы произошла реакции. Эта величина и называется энергией активации. При протекании реакции в обратном направлении энергия активации уже равна Е2. Именно такую дополнительную энергию должна приобрести система, находящаяся на уровне II для преодоления энергетического барьера. После этого образуются вещества, энергия которых отвечает уровню I. Представление об энергетическом барьере предполагает, что молекулы реагентов не всегда могут вступать в реакцию. Таким молекулам недостаточно просто встретиться. Они должны столкнуться, обладая при этом достаточно высокой кинетической энергией.
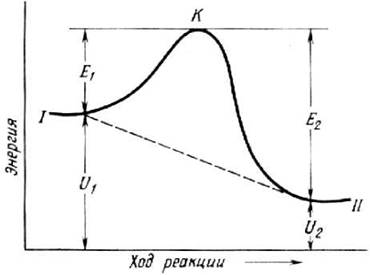
Рис. 5.3. Энергия состояния в ходе химической реакции и энергия активации [11]
За счет кинетической энергии этого движения и преодолевается энергетический барьер. По отношению к мономолекулярной фотодиссоциации это можно представить как то, что дополнительная энергия может быть получена молекулами реагентов за счет поглощения энергии излучения. Представления о том, что в ходе химической реакции система должна преодолеть энергетический барьер и что лишь некоторые «активные» молекулы могут вступать в химическое взаимодействие, подтверждаются большим количеством опытных фактов.
Возможность протекания химических реакций как в прямом, так и обратном направлении вполне согласуется с существованием энергетического барьера и трудно объясняется простым переходом непосредственно с энергетического уровня исходных веществ на уровень продуктов реакции (см. рис. 5.3, пунктирная линия). В случае очень большой энергии активации очень мало молекул, способных преодолеть энергетический барьер, и скорость реакции должна быть незначительной. Очень медленные реакции объясняются тем, что не все столкновения между молекулами реагентов ведут к химическим последствиям. Подсчеты показывают, что если бы все столкновения молекул были активными, то всякая химическая реакция имела бы взрывной характер. И, наконец, сильная зависимость скорости химических реакций от температуры тоже объясняется лишь в предположении о взаимодействии активных молекул, так как общее число столкновений не слишком сильно увеличивается с ростом температуры.
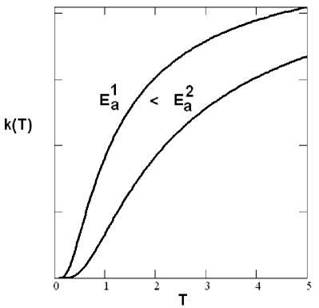
Рис. 5.4. Зависимость скорости реакции k от температуры T при различных энергиях активации
Равновесная лазерная химия. Таким образом, мы получили необходимые средства для моделирования распределения температуры в веществе при лазерном воздействии и описания протекания равновесных химических реакций во времени. Этого оказывается достаточно, чтобы объяснить очень большой объем экспериментальных результатов по тепловой лазерной модификации химической структуры вещества, полученных в 70 - 90 годы XX в.
Как видим, лазерное стимулирование равновесных (тепловых) реакций диссоциации все-таки существенно отличается от случая простого нагрева. Импульсное лазерное излучение позволяет очень быстро нагреть выбранные локализованные области пространства до высоких температур при небольшом среднем нагреве. Понятно, что в силу довольно резкой вначале зависимости выхода реакции от температуры (5.38), в сильно нагретых областях будут разрываться даже относительно высокоэнергетичные связи, обеспечивая продукты диссоциации, недостижимые при том же среднем по пространству и времени однородном нагреве. Так, даже при обычном гауссовом распределении интенсивности излучения в лазерном пучке будут наблюдаться продукты диссоциации связей с различными энергиями активации в различных точках пятна фокусировки (рис. 5.5).
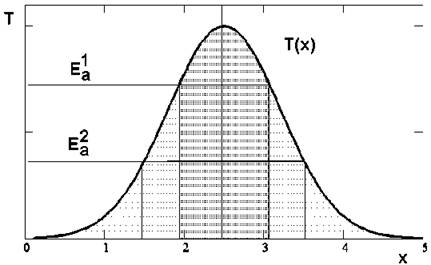
Рис. 5.5. Пояснение пространственной локализации равновесных процессов диссоциации в лазерном пучке
Легко видеть, что в случае однородной смеси молекул при тепловой диссоциации нет ни внутримолекулярной, ни межмолекулярной селективности воздействия. Однако и теперь лазерное излучение все же обеспечивает определенную селективность. Избирательность (селективность) в этом случае носит пространствен-
ный характер. Пространственная локализация энергии лазерного возбуждения определяется только коэффициентом поглощения и пространственным распределением интенсивности излучения. Тем самым, создавая заданные распределения интенсивности излучения в образце с известным распределением коэффициента поглощения, можно вызывать нужные процессы диссоциации молекул только в заданных пространственных областях.